
Наследствена остеодистрофия на Олбрайт. Псевдохипопаратироидизъм (наследствена остеодистрофия на Олбрайт): трудности при диференциално диагностично търсене. Клинично наблюдение Симптоми на псевдохипопаратироидизъм
Псевдохипопаратироидизъм
Какво е псевдохипопаратироидизъм -
Псевдохипопаратироидизъм(Гръцки pseudē s фалшив + хипопаратироидизъм; синоним: наследствена остеодистрофия на Олбрайт, болест на Олбрайт) е рядко наследствено заболяване скелетна система, симулиращ хипопаратиреоидизъм и характеризиращ се с нарушен метаболизъм на калций и фосфор; често придружени от забавено умствено и физическо развитие.
Какво провокира / Причини за псевдохипопаратироидизъм:
Причината за псевдохипопаратироидизма е вроден дефект - нечувствителност на периферните тъкани към действието на ПТХ.
Патогенеза (какво се случва?) по време на псевдохипопаратироидизъм:
Смята се, че псевдохипопаратироидизмът се основава на генетично обусловена резистентност на бъбреците и скелета към действието на паратиреоидния хормон в резултат на дефект в специфичния комплекс циторецептор - паратиреоиден хормон - аденилат циклаза, който нарушава образуването в бъбреците на цикличен 3 ", 5"-AMP, който е вътреклетъчен медиатор на действието на паратироидния хормон върху метаболитните процеси. Псевдохипопаратироидизмът е генетично хетерогенно заболяване. При някои пациенти самият циторецептор, който свързва паратироидния хормон, е дефектен (псевдохипопаратиреоидизъм тип Ia); други имат дефект в нуклеотид-свързващия протеин, локализиран в липидния двоен слой на клетъчната мембрана и функционално свързващ рецептора с аденилатциклазата (тип Ib). псевдохипопаратироидизъм). Някои пациенти изпитват ензимен дефицитсамата аденилат циклаза (псевдохипопаратироидизъм тип II). Дефицитът на cAMP в резултат на тези дефекти води до нарушаване на синтеза на специфични протеини, които определят биологичния ефект на паратироидния хормон. По този начин се губи чувствителността на целевите органи, по-специално на бъбреците, към паратироидния хормон. В резултат на това отделянето на фосфор с урината намалява, възниква хиперфосфатемия и вторично се развива хипокалцемия. Тъй като при псевдохипопаратироидизма паращитовидните жлези са непокътнати, може да се развие вторична в отговор на хипокалциемия, която стимулира производството на паратироиден хормон. хиперпаратироидизъм. Повишеното образуване на паратироиден хормон не води до увеличаване на екскрецията на фосфор и сАМР в урината поради генетично обусловената резистентност на бъбречните тубули към паратироидния хормон, но е придружено от промени в костна тъкан, характерен за хиперпаратироидизъм, което показва запазването на нормалната чувствителност на остеокластите към паратироидния хормон. При активност на псевдохипопаратиреоидизъм алкална фосфатазав кръвния серум е повишена или в нормални граници (0,5-1,3 µmolнеорганичен фосфор на 1 млкръвен серум за 1 чинкубиране при 37°; определение по Бодански). Всички варианти на псевдохипопаратиреоидизъм са наследствено заболяване, естеството на наследството е автозомно доминантно. Ниската плодовитост на мъжете, страдащи от псевдохипопаратироидизъм, обяснява рядкото предаване от баща на син; жените боледуват 2 пъти по-често от мъжете.
Обикновено при псевдохипопаратироидизъм се открива компенсаторна хиперплазия паращитовидни жлези(наличието на аденоми в тях не е характерно). В костната тъкан се наблюдават промени, типични за хиперпаратиреоидизъм - дифузна остеопороза, поява на кисти (т.нар. кафяви тумори, гигантски клетъчни тумори). Калцият, освободен от костите, се отлага под формата на калцификации в подкожна тъкан, както и в бъбреците, мускулите, миокарда, стените големи артерии, конюнктивата на окото и по периферията на роговицата.
Симптоми на псевдохипопаратироидизъм:
Клиничните признаци на псевдохипопаратироидизъм са подобни на тези на идиопатичния хипопаратироидизъм. Има атаки на тонични конвулсии, които възникват спонтанно или под въздействието на някакви дразнители. Калцификатите в подкожната тъкан са склонни към разязвяване. Подкожната осификация често е тежка до степен, че имитира осифициращия миозит. Характерните особености включват умствена изостаналост, забавен растеж, лунообразно лице, затлъстяване и брахидактилия, особено скъсяване на първия, четвъртия и петия метакарпал и метатарзал. Могат да се наблюдават множество екзостози, дисхондроплазия, прояви вторичен хиперпаратироидизъмпод формата на субпериостална резорбция на костите на пръстите; промените в епифизите на костите са същите като при фиброзна остеодисплазия. Често се отбелязва повръщане, както и хематурия поради образуването на оксалатни камъни в пикочните пътища, откриват се лещовидна катаракта и хипоплазия на зъбния емайл.
При пациенти с псевдохипопаратиреоидизъм, заедно с намаляване на чувствителността на целевите органи към паратироидния хормон, може да се наблюдава резистентност към други хормони, зависими от аденилатциклазната система, например половите жлези към гонадотропните хормони, щитовидната жлеза към тиреостимулиращия хормон , насочват органите към глюкагон и антидиуретичен хормон. Има повишена честота автоимунни заболяванияИ захарен диабет, се наблюдава хипотиреоидизъм и хипертиреоидизъм.
Съществува и псевдопсевдохипопаратироидизъм, който се характеризира с липсата на хипокалциемия, хиперфосфатемия, гърчове и остеомалация.
Диагностика на псевдохипопаратироидизъм:
Диагнозата в типичните случаи на заболяването се поставя при деца на възраст 5-10 години въз основа на характерна клинична картина, множество аномалии в развитието на костния скелет, наличие на хипокалцемия, хиперфосфатемия, нормална или повишена активност на алкалната фосфатаза. в кръвния серум, намалена екскреция на калций и фосфор в урината, високо съдържаниепаратиреоиден хормон в кръвта. Наличието на резистентност на бъбречните тубули към паратироидния хормон се потвърждава чрез тест, базиран на определяне на количеството фосфат и сАМР, екскретирани в урината. Липсата на значително увеличение на съдържанието на фосфати и сАМР в урината след прилагане на паратироиден хормон на пациента показва бъбречна резистентност към действието на паратироидния хормон. При пациенти с идиопатичен и следоперативен хипопаратиреоидизъм, напротив, след интравенозно приложение на 200 единици паратироиден хормон в урината, съдържанието на фосфати и сАМР в рамките на 4 чсе увеличава 2-10 пъти в сравнение с базова линия. Екскрецията на хидроксипролин в урината при нелекувани пациенти с псевдохипопаратироидизъм е нормална или леко повишена, а при хипопаратироидизъм е намалена. Рентгеновата диагностика на псевдохипопаратироидизма се основава на идентифициране на специфични промени в костите и меките тъкани.
Псевдохипопаратироидизмът в комбинация с хипогонадизъм при жените трябва да се разграничава от Синдром на Шерешевски-Търнър, на които псевдопсевдохипопаратироидизмът е фенотипно подобен. При синдрома на Шерешевски-Търнър липсва полов хроматин, на мястото на яйчниците се намират нишки от съединителна тъкан, които не се откриват при ректално и ултразвуково изследване.
Лечение на псевдохипопаратироидизъм:
Лечението на хипокалцемия се състои в предписване на калциеви добавки в дози, достатъчни за поддържане на нормални концентрации на калций в кръвта. Голямо значениеима терапия с витамин D. Началната доза се изчислява от 2000 IU/ килограмателесно тегло на ден, но не повече от 100 000 IU на ден. За да се избегне предозиране на препарати с витамин D, е необходимо да се следи концентрацията на калций в кръвта на всеки 3-7 дни през първите две седмици от лечението и всеки месец през следващите 2-3 месеца. След достигане на стабилна концентрация на калций в кръвта е достатъчно да се проверява веднъж на 2-3 месеца. Можете да използвате калцитрин, дихидротахистерол, оксидевит, както и други лекарства активни формивитамин D. Диета с ограничен прием на фосфор спомага за нормализиране на концентрацията на калций в кръвта и премахване на симптомите на вторичен хиперпаратироидизъм. При недостатъчност на други жлези вътрешна секрецияпровеждайте заместителна терапия с подходящи хормони. Лечението с паратироиден хормон не е ефективно. За спиране на конвулсивните атаки се прилага интравенозно 10% разтвор на калциев хлорид или калциев глюконат; перорално - 5-10% разтвор на калциев хлорид, 1 супена лъжица 3-4 пъти на ден: калциев глюконат, калциев лактат - до 10 Жв един ден.
Прогнозата с рационална терапия е благоприятна. Като се има предвид наследствената природа на псевдохипопаратироидизма, е необходимо медицинско генетично консултиране относно възможността за псевдохипопаратироидизъм в потомството.
Профилактика на псевдохипопаратироидизъм:
Към кои лекари трябва да се обърнете, ако имате псевдохипопаратироидизъм:
Притеснява ли те нещо? Искате ли да научите по-подробна информация за псевдохипопаратироидизма, неговите причини, симптоми, методи на лечение и профилактика, хода на заболяването и диетата след него? Или имате нужда от преглед? Можеш запишете си час при лекар– клиника евролабораториявинаги на ваше разположение! Най-добрите лекари ще ви прегледат и проучат външни признации ще ви помогне да идентифицирате заболяването по симптоми, ще ви посъветва и ще ви окаже необходимата помощ и ще постави диагноза. вие също можете обадете се на лекар у дома. Клиника евролабораторияотворен за вас денонощно.
Как да се свържете с клиниката:
Телефонен номер на нашата клиника в Киев: (+38 044) 206-20-00 (многоканален). Секретарят на клиниката ще избере удобен ден и час за посещение при лекаря. Нашите координати и посоки са посочени. Разгледайте по-подробно всички услуги на клиниката по него.
(+38 044) 206-20-00
Ако преди това сте правили някакви изследвания, Не забравяйте да занесете резултатите от тях на лекар за консултация.Ако изследванията не са направени, ние ще направим всичко необходимо в нашата клиника или с наши колеги в други клиники.
Вие? Необходимо е да се подходи много внимателно към цялостното ви здраве. Хората не обръщат достатъчно внимание симптоми на заболяванияи не осъзнават, че тези заболявания могат да бъдат животозастрашаващи. Има много заболявания, които в началото не се проявяват в тялото ни, но накрая се оказва, че за съжаление вече е късно да се лекуват. Всяко заболяване има свои специфични симптоми, характерни външни прояви- т.нар симптоми на заболяването. Идентифицирането на симптомите е първата стъпка в диагностицирането на заболявания като цяло. За да направите това, просто трябва да го правите няколко пъти в годината. бъдете прегледани от лекарне само да се предотврати ужасна болест, но и за поддържане на здрав дух в тялото и организма като цяло.
Ако искате да зададете въпрос на лекар, използвайте секцията за онлайн консултация, може би там ще намерите отговори на вашите въпроси и ще прочетете съвети за грижа за себе си. Ако се интересувате от отзиви за клиники и лекари, опитайте се да намерите необходимата информация в раздела. Регистрирайте се и на медицинския портал евролабораторияза да сте в крак с времето последни новинии актуализации на информация на уебсайта, които автоматично ще ви бъдат изпращани по имейл.
Други заболявания от групата Болести на ендокринната система, разстройства на храненето и обмяната на веществата:
| Адисонова криза (остра надбъбречна недостатъчност) |
| Аденом на гърдата |
| Адипозогенитална дистрофия (болест на Perchkranz-Babinski-Fröhlich) |
| Адреногенитален синдром |
| акромегалия |
| Хранителна лудост (хранителна дистрофия) |
| Алкалоза |
| Алкаптонурия |
| Амилоидоза (амилоидна дистрофия) |
| Амилоидоза на стомаха |
| Амилоидоза на червата |
| Амилоидоза на панкреатични острови |
| Чернодробна амилоидоза |
| Амилоидоза на хранопровода |
| ацидоза |
| Белтъчно-енергийно недохранване |
| I-клетъчна болест (муколипидоза тип II) |
| Болест на Уилсън-Коновалов (хепатоцеребрална дистрофия) |
| Болест на Гоше (глюкоцереброзидна липидоза, глюкоцереброзидоза) |
| Болест на Иценко-Кушинг |
| Болест на Krabbe (глобоидна клетъчна левкодистрофия) |
| Болест на Ниман-Пик (сфингомиелиноза) |
| Болест на Фабри |
| Ганглиозидоза GM1 тип I |
| Ганглиозидоза GM1 тип II |
| Ганглиозидоза GM1 тип III |
| Ганглиозидоза GM2 |
| Ганглиозидоза GM2 тип I (амавротичен идиотизъм на Тей-Сакс, болест на Тей-Сакс) |
| GM2 ганглиозидоза тип II (болест на Sandhoff, амавротичен идиотизъм на Sandhoff) |
| Ювенилна ганглиозидоза GM2 |
| Гигантизъм |
| Хипералдостеронизъм |
| Вторичен хипералдостеронизъм |
| Първичен хипералдостеронизъм (синдром на Conn) |
| Хипервитаминоза D |
| Хипервитаминоза А |
| Хипервитаминоза Е |
| Хиперволемия |
| Хипергликемична (диабетна) кома |
| Хиперкалиемия |
| Хиперкалциемия |
| Хиперлипопротеинемия тип I |
| Хиперлипопротеинемия тип II |
| Хиперлипопротеинемия тип III |
| Хиперлипопротеинемия тип IV |
| Хиперлипопротеинемия тип V |
| Хиперосмоларна кома |
| Вторичен хиперпаратироидизъм |
| Първичен хиперпаратироидизъм |
| Хиперплазия на тимуса (тимусната жлеза) |
| Хиперпролактинемия |
| Хиперфункция на тестисите |
| Хиперхолестеролемия |
| хиповолемия |
| Хипогликемична кома |
| Хипогонадизъм |
| Хипогонадизъм хиперпролактинемия |
| Изолиран хипогонадизъм (идиопатичен) |
| Първичен вроден хипогонадизъм (анорхизъм) |
| Първичен придобит хипогонадизъм |
| Хипокалиемия |
| Хипопаратироидизъм |
| Хипопитуитаризъм |
| Хипотиреоидизъм |
| Гликогеноза тип 0 (агликогеноза) |
| Гликогеноза тип I (болест на Gierke) |
| Гликогеноза тип II (болест на Помпе) |
| Гликогеноза тип III (болест на морбили, болест на Форбс, лимитирана декстриноза) |
| Гликогеноза тип IV (болест на Андерсен, амилопектиноза, дифузна гликогеноза с чернодробна цироза) |
| Гликогеноза тип IX (болест на Haga) |
| Гликогеноза тип V (болест на McArdle, миофосфорилазен дефицит) |
| Гликогеноза тип VI (болест на Херс, дефицит на хепатофосфорилаза) |
| Гликогеноза тип VII (болест на Таруи, дефицит на миофосфофруктокиназа) |
| Гликогеноза тип VIII (болест на Томсън) |
| Гликогеноза тип XI |
| Гликогеноза тип X |
| Дефицит (недостатъчност) на ванадий |
| Магнезиев дефицит (недостатъчност) |
| Дефицит на манган (недостатъчност) |
| Дефицит на мед (недостатъчност) |
| Дефицит (недостатъчност) на молибден |
| Дефицит (недостатъчност) на хром |
| Дефицит на желязо |
| Дефицит на калций (хранителен дефицит на калций) |
| Дефицит на цинк (диетичен дефицит на цинк) |
| Диабетна кетоацидотична кома |
| Дисфункция на яйчниците |
| Дифузна (ендемична) гуша |
| Забавен пубертет |
| Излишък на естроген |
| Инволюция на млечните жлези |
| Нанизъм (нисък ръст) |
| Квашиоркор |
| Кистозна мастопатия |
| Ксантинурия |
| Млечно-ацидемична кома |
| Левциноза (болест от кленов сироп) |
| Липидози |
| Липогрануломатоза на Фарбер |
| Липодистрофия (мастна дегенерация) |
| Вродена генерализирана липодистрофия (синдром на Seyp-Lawrence) |
| Хипермускулна липодистрофия |
| Липодистрофия след инжектиране |
| Прогресивна сегментна липодистрофия |
| Липоматоза |
| Липоматозата е болезнена |
| Метахроматична левкодистрофия |
| Микседемна кома |
Псевдохипопаратироидизъм - достатъчно рядко заболяванескелетна система, чиято същност е нарушение на метаболизма на калций и фосфор. Пациентът обикновено изпитва инхибиране на умственото и физическото развитие. Заболяването е наследствено.
От факта, че името на заболяването има представката "псевдо", лесно може да се разбере, че то имитира хипопаратироидизъм. Второто име на това заболяване е наследствената остеодистрофия на Олбрайт, кръстена на лекаря, който изучава и описва тази патология в средата на миналия век.
Видове псевдохипопаратироидизъм
Има два вида псевдохипопаратироидизъм - в зависимост от това дали нивото на калций в кръвта се е променило или не. Първият тип болест на Олбрайт има същото Клинични признаци, както при идиопатичния хипопаратироидизъм, но се характеризира с намаляване на нивата на калций в кръвта. Няма тъканна чувствителност към паратироидния хормон. Вторият тип псевдохипопаратироидизъм, напротив, се различава по това, че нивото на калций е нормално. Следователно този тип се нарича псевдопсевдохипопаратироидизъм. Между другото, според ендокринолозите, една форма може лесно да премине в друга и членовете на едно и също семейство могат да имат различни видовезаболявания.
Причини за псевдохипопаратироидизъм
Нарушенията на фосфорно-калциевия метаболизъм се развиват поради резистентност на тъканите към паратироидния хормон, който се произвежда от паращитовидните жлези. Въз основа на псевдохипопаратиреоидизъм за първи път се потвърждава феноменът на нарушена чувствителност на тъканите към хормона, произвеждан от жлезите с вътрешна секреция или въведен външно.
Псевдохипопаратироидизмът е генетична патология. Причинява се от вроден синдром - специфичен клетъчен рецептор - паратиреоиден хормон-аденилат циклаза, поради което периферните тъкани губят чувствителност към паратироидния хормон.
Кой е най-податлив на псевдохипопаратироидизъм? На първо място, децата на пациента и други роднини, тъй като заболяването е наследствено, автозомно доминантно. Жените страдат по-често от болестта на Олбрайт. Експертите обясняват това с факта, че мъжете с псевдохипопаратироидизъм имат ниска плодовитост, а самата наследственост се предава от баща на син само в отделни случаи.
Няма статистика за това колко често е това заболяване, около триста случая са описани в медицински трудове.
Симптоми на псевдохипопаратироидизъм
- Органни проблеми ендокринна системас псевдохипопаратироидизъм се определят и основните симптоми: пациентът обикновено е набит, по-нисък от другите, поради скъсяване долните крайници, страда от затлъстяване, нивата на кръвната захар са повишени, лицето придобива лунообразна форма;
- Засегнати са представителките на нежния пол менструален цикъл. Често тези признаци са придружени от акромегалия, гинекомастия, различни автоимунни процеси, синдром на Иценко-Кушинг и други заболявания. При такива пациенти рискът от захарен диабет и безвкусен диабет също е по-висок;
- Страдащите от болестта на Олбрайт често се оплакват от проблеми със зъбите (емайлът им е увреден), пациентите също се притесняват от повръщане, тонични гърчове - както спонтанни, така и под въздействието на дразнители, може да се наблюдава урина в кръвта. Съществува риск от катаракта. Характеризира се с умствена изостаналост. Децата с това заболяване не могат да се справят с училищната програма, паметта им е намалена;
- IN костно-ставна системанастъпват и значителни промени - появява се дифузна остеопороза, развиват се кисти (кафяви тумори). Пациентите с псевдохипопаратироидизъм са по-склонни да страдат от фрактури или костни деформации. Техните първа, четвърта и пета метакарпална и метатарзална кост са скъсени, а по ръцете и краката се наблюдават признаци на костна резорбция. В допълнение, калцият, който се освобождава от костите, започва да се натрупва в подкожната тъкан, образувайки калцификации (това се случва и при осифициращ миозит), в областта на ставите и черепната кухина. Калцификации има и в мускулите, бъбреците, миокарда и по стените на големите артерии;
- Друг отличителен белегБолест на Олбрайт - пигментация на кожата. Кафяви петна се разпространяват по тялото (както от едната, така и от другата страна).
По отношение на работоспособността зависи от тежестта на процеса и ефективността лечение с лекарства. Ако формата на заболяването е латентна и няма очевидни атаки, тогава способността за работа е частично запазена. Но има редица ограничения - по-специално не можете да работите с движещи се механизми или в транспорта; невропсихическото пренапрежение и прекомерният физически труд също са забранени. Ако има чести конвулсивни атаки, умствена изостаналост е ясно изразена или значително зрително увреждане поради катаракта, тогава такива пациенти вече се считат за инвалиди.
Диагностика на псевдохипопаратироидизъм
Симптомите са доста специфични и ако започнат да се появяват, трябва незабавно да се свържете с квалифициран лекар. Това е ранното и правилна диагнозагарантира успеха на лечението.
Тъй като болестта на Олбрайт е вродена, тя обикновено се диагностицира на пациента в детска възраст (предучилищна, младша възраст). училищна възраст). Генетичните лекари правят заключения въз основа на клиничните симптоми, използват се и лабораторни и инструментални диагностични методи.
По-специално, за да се изясни диагнозата, пациентът дарява кръв (при пациенти с псевдохипопаратиреоидизъм нивото на калций е ниско, а фосфорът и паратироидният хормон са високи, активността на алкалната фосфатаза в кръвта е висока), урина (при пациента това анализът ще покаже намаляване на екскретирания фосфор и калций). Правят се и специални тестове, които показват колко чувствителна е бъбречната тубулна тъкан на пациента към паратироидния хормон. Понякога става необходимо да се оцени нивото на хидроксипролин.
Инструменталните методи включват рентгенова диагностика (тя е най-информативна при идентифициране на специфични промени в костната и мускулната тъкан).
Лечение на псевдохипопаратироидизъм
Тази рядка наследствена патология се лекува с калциеви добавки. Лекарят избира дозата въз основа на съдържанието на калций, необходимо за поддържане на хомеостазата. И за да може калцият да се усвоява безпроблемно, в терапевтичния режим са включени и препарати от витамин D. Освен това лекарят за пациент с псевдохипопаратиреоидизъм ще разработи диета, при която ще бъде ограничено количеството храни, съдържащи флуор. Тази диета ще помогне за нормализиране на концентрацията на калций в кръвта и ще премахне признаците на вторичен хиперпаратироидизъм.
Ако болестта на Олбрайт е придружена от други ендокринни нарушения, тогава може да има нужда от хормонална заместителна терапия. Конвулсиите се облекчават с разтвор на калциев хлорид или калциев глюконат, който се прилага интравенозно. Ако се появи катаракта, тя се лекува от офталмолог, а психологът помага за облекчаване на когнитивното увреждане.
При добре обмислен режим на лечение прогнозата е много благоприятна. Но всички, които са имали псевдохипопаратироидизъм, трябва редовно да се консултират с генетици, за да предотвратят заболяването при своите потомци.
Профилактика на псевдохипопаратироидизъм
Като се има предвид, че болестта е наследствена, тогава единствения начинпревенцията за тези, които имат фамилна анамнеза за псевдохипопаратиреоидизъм, е редовното посещение на медицински генетици за консултации.
(болест на Олбрайт) е наследствена остеодистрофия, причинена от резистентност на периферните тъкани към паратироидния хормон, която е придружена от нарушение на калциево-фосфорния метаболизъм, забавено физическо и умствено развитие. Псевдохипопаратироидизмът протича със симптоми на дифузна остеопороза, тонични гърчове, фрактури и деформации на костите, отлагане на калций в мускулите и кръвоносните съдове, образуване на камъни в пикочните пътища, забавяне на растежа и умствена изостаналост. За диагностициране на псевдохипопаратироидизъм се определя нивото на калций, алкална фосфатаза и паратиреоиден хормон в кръвта; отделяне на фосфор и калций с урината; функционален тестс въвеждането на паратиреоиден хормон; рентгенова диагностика. Лечението на псевдохипопаратиреоидизма включва медикаментозна корекция на хипокалциемията.
МКБ-10
E20.1

Главна информация
Псевдохипопаратироидизмът е рядка наследствена патология; В ендокринологията са известни само около 300 случая на това заболяване. Псевдохипопаратиреоидизмът по своите прояви прилича на хипопаратиреоидизъм. Въпреки това, ако хипопаратироидизмът се основава на първичен дефицит на паратироиден хормон, причинен от намаляване на функционалната активност на паращитовидните жлези, тогава при болестта на Олбрайт псевдохипопаратироидният синдром се причинява от нарушение на чувствителността на целевите тъкани към действието на паращитовидните жлези. хормон с достатъчно ниво на секреция.
Псевдохипопаратироидизмът е описан за първи път през 1942 г. Ф. Олбрайт и според автора се нарича „болест на Олбрайт” или „наследствена остеодистрофия на Олбрайт”. Първият вариант на заболяването се характеризира с аномалии в развитието на скелета, резистентност на периферните тъкани към паратироидния хормон, хипокалцемия и клинична картина, подобно на идиопатичния хипопаратироидизъм. Вторият вариант на болестта на Олбрайт протича според нормокалцемичния тип и в литературата се нарича псевдопсевдохипопаратироидизъм. Описани са случаи на развитие в едно семейство както на нормо-, така и на хипокалциемични варианти на заболяването, както и на прехода на една форма в друга.
Причини за псевдохипопаратироидизъм
При всеки вариант на псевдохипопаратиреоидизъм заболяването е наследствено по природа с автозомно-доминантен тип наследство, свързано с Х-хромозомата. Проучване на родословията показва, че броят на жените с псевдохипопаратироидизъм е 2 пъти по-висок от броя на болните мъже; Освен това болестта на Олбрайт не се предава от баща на син.
Псевдохипопаратироидизмът се причинява от генетична резистентност на скелета и бъбреците към действието на паратироидния хормон. Това се дължи на дефект в специфични рецептори на плазмените мембрани на целевите клетки и дефицит на ензимите аденилат циклаза, протеин киназа, цикличен 3,5-аденозин монофосфат (AMP) в дисталните тубули на нефрона, което е придружено от реабсорбция на фосфор. Намалената екскреция на фосфор в урината води до хиперфосфатемия и вторична хипокалцемия.
Тъй като при псевдохипопаратиреоидизъм паращитовидните жлези остават интактни, хипокалциемията може да предизвика стимулиране на секрецията на паратиреоиден хормон и развитие на вторичен хиперпаратиреоидизъм. При псевдохипопаратиреоидизъм обикновено има компенсаторна хиперплазия на паращитовидните жлези (развитието на аденоми не е типично), промени в костната тъкан (остеопороза, кисти), отлагане на калцификации в скелетни мускули, подкожната тъкан, както и в бъбреците, артериалните стени, миокарда, конюнктивата и роговицата. Псевдохипопаратироидизмът често се комбинира с артериална хипертония, захарен диабет, артериит и полиартрит.
Симптоми на псевдохипопаратироидизъм
Клиничните прояви на псевдохипопаратиреоидизма са подобни на тези на идиопатичния хипопаратиреоидизъм. Ендокринните симптоми на псевдохипопаратироидизъм включват нисък ръст, затлъстяване и хипергликемия, лунно лице и олигоменорея. Болестта на Олбрайт често се комбинира с други ендокринни заболявания - акромегалия, синдром на Кушинг, гинекомастия, хипотиреоидизъм или хипертиреоидизъм.
Остеоартикуларните признаци на псевдохипопаратироидизъм включват брахидактилия с изразено скъсяване на 1-ви, 4-ти и 5-ти метакарпални и метатарзални кости; екзостози, дисхондроплазия, промени в епифизните краища на костите, резорбция на костите на пръстите. Характеризира се със забавено никнене на зъби, хипоплазия на зъбния емайл, подкожна осификация.
Неврологичните симптоми на псевдохипопаратироидизъм са представени от атаки на тонични конвулсии, които могат да възникнат спонтанно или под въздействието на всякакви дразнители. За пациентите с псевдохипопаратиреоидизъм е характерна умствена изостаналост. Други симптоми на псевдохипопаратироидизъм включват уролитиаза, хематурия, повръщане и развитие на лещовидна катаракта. При псевдопсевдохипопаратироидизъм няма хипокалцемия, хиперфосфатемия, остеомалация и конвулсивен синдром.
Диагностика на псевдохипопаратироидизъм
В типичните случаи псевдохипопаратироидизмът се диагностицира при деца на възраст 5-10 години, като се вземат предвид характерните симптоми, лабораторните и радиологичните данни. При пациенти с псевдохипопаратиреоидизъм се определят хипокалциемия, хиперфосфатемия, нормална или повишена активност на алкалната фосфатаза в кръвта и повишени нива на паратироидния хормон в кръвния серум; намалена екскреция на калций и фосфор в урината.
Нечувствителността на бъбречните тубули към паратироидния хормон се потвърждава чрез тест, базиран на определяне на количеството фосфат и цикличен аденозин монофосфат, екскретиран в урината. При псевдохипопаратироидизъм в отговор на венозно приложениепаратироиден хормон, няма значително увеличение на съдържанието на фосфати и сАМР в урината.
В случай на псевдохипопаратироидизъм е необходима корекция на хипокалциемията, поради което лечението с калциеви препарати се провежда в дози, които позволяват поддържане на нормални концентрации на калций в кръвта. Приемът на витамин D и неговите активни форми е показан под контрола на концентрацията на калций в кръвния серум. Лечението с паратироиден хормон не е ефективно. За нормализиране на концентрацията на калций и премахване на симптомите на вторичен хиперпаратироидизъм е необходимо да се спазва диета с ограничен прием на фосфор.
При функционална недостатъчност на други жлези се провежда заместителна терапия с подходящи хормони. Ако се появят гърчове, е показано интравенозно приложение на калциеви разтвори.
Навременната диагноза и рационалната терапия на псевдохипопаратироидизма ни позволява да говорим за положителни прогнози за живота и възможността за контролиране на хода на заболяването. Като се има предвид генетичната природа на псевдохипопаратироидизма, препоръчително е да се проведе медицинско генетично консултиране, за да се оцени рискът от болестта на Олбрайт в потомството.
![]()
Описание:
Псевдохипопаратиреоидизмът (на гръцки pseudēs фалшив +; синоним: наследствена остеодистрофия на Олбрайт, болест на Олбрайт) е рядко наследствено заболяване на скелетната система, симулиращо хипопаратиреоидизъм и характеризиращо се с нарушен метаболизъм на калций и фосфор; често придружени от забавено умствено и физическо развитие.
Симптоми:
Клиничните признаци на псевдохипопаратиреоидизма са подобни на тези на идиопатичния хипопаратиреоидизъм. Има тонични атаки, които възникват спонтанно или под въздействието на всякакви дразнители. Калцификатите в подкожната тъкан са склонни към разязвяване. Подкожната осификация често е тежка до степен, че имитира осификация. Характеризира се с умствена изостаналост, забавяне на растежа, лунообразно лице и особено скъсяване на първата, четвъртата и петата метакарпална и метатарзална кост. Могат да се наблюдават множество екзостози, дисхондроплазия и вторични прояви под формата на субпериостална резорбция на костите на пръстите; промените в епифизите на костите са същите като при фиброзна остеодисплазия. Често се отбелязва повръщане, както и хематурия поради образуването на оксалатни камъни в пикочните пътища, откриват се лещовидна катаракта и хипоплазия на зъбния емайл.
При пациенти с псевдохипопаратиреоидизъм, заедно с намаляване на чувствителността на целевите органи към паратироидния хормон, може да се наблюдава резистентност към други хормони, зависими от аденилатциклазната система, например половите жлези към гонадотропните хормони, щитовидната жлеза към тиреостимулиращия хормон , насочват органите към глюкагон и антидиуретичен хормон. Наблюдава се повишена заболеваемост от автоимунни заболявания и захарен диабет, като се наблюдават.
Съществува и псевдопсевдохипопаратироидизъм, който се характеризира с липса на гърчове и остеомалация.
Причини:
Причината за псевдохипопаратироидизма е вроден дефект - нечувствителност на периферните тъкани към действието на ПТХ.
Лечение:
За лечение се предписва следното:
Лечението на хипокалцемия се състои в предписване на калциеви добавки в дози, достатъчни за поддържане на нормални концентрации на калций в кръвта. От голямо значение е терапията с витамин D. Началната доза се изчислява от 2000 IU/kg телесно тегло на ден, но не повече от 100 000 IU на ден. За да се избегне предозиране на препарати с витамин D, е необходимо да се следи концентрацията на калций в кръвта на всеки 3-7 дни през първите две седмици от лечението и всеки месец през следващите 2-3 месеца. След достигане на стабилна концентрация на калций в кръвта е достатъчно да се проверява веднъж на 2-3 месеца. Можете да използвате калцитрин, дихидротахистерол, оксидевит, както и други препарати от активни форми на витамин D. Диета с ограничаване на фосфора помага за нормализиране на концентрацията на калций в кръвта и премахване на симптомите на вторичен хиперпаратироидизъм. При недостатъчност на други ендокринни жлези се провежда заместителна терапия с подходящи хормони. Лечението с паратироиден хормон не е ефективно. За спиране на конвулсивните атаки се прилага интравенозно 10% разтвор на калциев хлорид или калциев глюконат; перорално - 5-10% разтвор на калциев хлорид, 1 супена лъжица 3-4 пъти на ден: калциев глюконат, калциев лактат - до 10 g на ден.
Прогнозата с рационална терапия е благоприятна. Като се има предвид наследствената природа на псевдохипопаратироидизма, е необходимо медицинско генетично консултиране относно възможността за псевдохипопаратироидизъм в потомството.
Е.В. Тозлиян, детски ендокринолог, генетик, д-р И.В. Шулякова, невролог, д-р,
изолиран структурно подразделение"Научно-изследователски клиничен институт по педиатрия" Държавна бюджетна образователна институция за висше професионално образование "Руски национален изследователски медицински университет на името на N.I. Пирогов" на Министерството на здравеопазването на Руската федерация, Москва
Ключови думи:
деца, псевдохипопаратиреоидизъм, наследствена остеодистрофия на Олбрайт, затлъстяване, хипокалциемия, диагноза, резистентност към паратхормон.
Ключови думи:деца, псевдохипопаратиреоидизъм, наследствена остеодистрофия на Олбрайт, затлъстяване, хипокалцемия, диагностика, резистентност към паратиреоиден хормон.
Псевдохипопаратиреоидизъм (на гръцки pseudes - фалшив + хипопаратиреоидизъм; синоним: наследствена остеодистрофия на Олбрайт, синдром на яванското пиле) е рядко наследствено заболяване на скелетната система, симулиращо хипопаратиреоидизъм и характеризиращо се с нарушен метаболизъм на калций и фосфор; често придружени от забавено умствено и физическо развитие. Заболяването е описано за първи път от американския ендокринолог Олбрайт Ф. през 1942 г. Разпространението на заболяването е 7,9 на 1 милион души.
ГЕНЕТИЧНИ ДАННИ
Псевдохипопаратироидизмът (PHP) е генетично хетерогенно заболяване. Данните за вида на наследственото предаване са противоречиви: както Х-свързан доминантен, така и автозомно-доминантен, автозомно-рецесивен тип. В повечето случаи развитието на наследствената остеодистрофия на Олбрайт се свързва с мутации в локуса 20q13 на гена GNAS1, разположен на хромозома 20 (Patten et al., 1990), кодиращ Gs-алфа протеина, свързан с рецептора на паратироидния хормон (PTH). . Подобен фенотип беше открит и при пациенти с интерстициална делеция на дългото рамо на хромозома 2 в локуса 2q37.
ПАТОГЕНЕЗА
Патогенезата на псевдохипопаратиреоидизма се основава на генетично обусловена резистентност на бъбреците и скелета към действието на паратиреоидния хормон в резултат на дефект в комплекса „специфичен циторецептор – паратиреоиден хормон – аденилатциклаза“, който нарушава образуването на цикличен 3"- , 5"-аденозин монофосфат (сАМР) в бъбреците, който е вътреклетъчен медиатор на действието на паратироидния хормон върху метаболитните процеси. Псевдохипопаратиреоидизмът е генетичен хетерогенно заболяване.При някои пациенти самият циторецептор, който свързва паратиреоидния хормон, е дефектен (тип 1А псевдохипопаратиреоидизъм); други имат дефект в нуклеотид-свързващия протеин, локализиран в липидния двоен слой на клетъчната мембрана и функционално свързващ рецептора с аденилат циклазата (тип 1В). псевдохипопаратироидизъм). Някои пациенти имат ензимен дефицит на самата аденилат циклаза (тип 2 псевдохипопаратироидизъм). Дефицитът на cAMP в резултат на тези дефекти води до нарушаване на синтеза на специфични протеини, които определят биологичния ефект на паратироидния хормон. По този начин се губи чувствителността на целевите органи към паратироидния хормон.
КЛИНИЧНА ХАРАКТЕРИСТИКА
В момента има 4 клинични формипатологии: типове 1A, 1B, 1C и 2. Познаване на техните клинични и биохимични характеристики и данни генетични изследваниядава възможност за диференциална диагноза в рамките на самата нозологична форма.
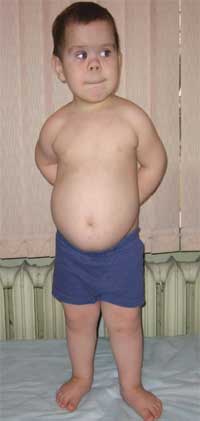
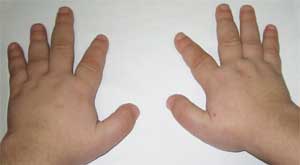
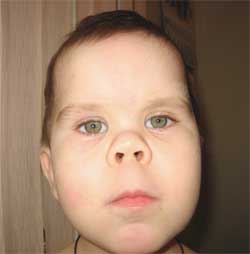
Честите признаци, които позволяват да се подозира заболяването, са диспропорционалност във физическото развитие, нисък ръст (до степен на нанизъм) поради скъсяване на долните крайници (снимка 1), брахидактилия (снимка 2) и кръгла „лунообразна форма“ лице (снимка 3). Понякога се наблюдават екзостози и зъбна аплазия.
Снимка 1.
Външен виддете с остеодистрофия на Олбрайт
(характеристики на фенотипа, нисък ръст поради скъсяване на долните крайници)
Снимка 2.
Характеристики на скелетната система на пациента
с остеодистрофия на Олбрайт
(брахидактилия - скъсяване на пръстите)
Снимка 3.
Характеристики на фенотипа на детето
с остеодистрофия на Олбрайт
(кръгло лице с форма на луна)
Патогномоничен признак е рязкото скъсяване на I, III и V метакарпални и метатарзални кости (особено III и IV), в резултат на което II пръсти на ръцете и краката са по-дълги от останалите и когато ръката е стисната в юмрук, няма изпъкналости в областта на IV и V метакарпофалангеални стави - т. нар. брахиметафалангизъм. Откриват се също къси широки фаланги, удебеляване на черепния свод и деминерализация на костите (остеопороза), затлъстяване.
Умствена изостаналост(обикновено умерена тежест) се открива при приблизително 20% от пациентите. Според някои автори умствената изостаналост се среща в 70% от случаите при наличие на хипокалциемия и в 30% от случаите при нормокалцемия. Психични процесипри пациентите се забавят. В неврологичния статус често се отбелязват двигателна неловкост и невротични реакции: страхове, тревожност, безпокойство, лош сън, повишени рефлекси, конвулсии с тетаничен характер и причинени от хипокалцемия, понякога конвулсивни пароксизми. Описани са и миопатични симптоми: мускулна умора, мускулна слабост. Често се наблюдават екстрапирамидни нарушения: хореиформна хиперкинеза, атетоза, лицев хемиспазъм, паркинсонизъм, в някои случаи има епилептични пароксизми, малкомозъчни симптоми: атаксия, загуба на координация.
Често се открива калцификация на меките тъкани, подкожни калцификации (гърди, корем, сухожилия на петата), с хистологично изследванекойто - остеома кутис(Izraeli et al., 1992), мозък (базални ганглии). Важно е да се отбележи, че калцификатите може вече да са налице при раждането. В резултат на хипокалцемия обикновено се развива катаракта и възникват дефекти на зъбния емайл.
ПСЕВДОХИПОПАРАТРОЗА ТИП 1А
има автозомно доминантен модел на наследяване. Генът за псевдохипопаратиреоидизъм тип 1А - GNAS1 - е локализиран на дългото рамо на хромозома 20, в локуса 20q13.2. Развитието на заболяването е свързано с дефицит на гуанин нуклеотид-свързващ протеин (Gs протеин). В същото време PTH, свързвайки се с рецепторите на целевите тъкани, не е в състояние да активира цикличния аденозин монофосфат (cAMP) и да предизвика тъканен отговор. Вероятно подобен механизъм е в основата на развитието на нечувствителност на тъканите на други органи и ендокринни жлези (хипофункция на щитовидната жлеза, половите жлези, хипофизната жлеза, диабет, както и намален отговор на черния дроб към прилагане на глюкагон), наблюдавани при псевдохипопаратироидизъм тип 1А. При този тип патология не се наблюдава нормалната повишена екскреция на сАМР в урината в отговор на екзогенно приложение на ПТХ. Заболяването се диагностицира по-често на възраст 5-10 години. Болните са с нисък ръст, къс врат, кръгло лице, скъсяване на метакарпалните и метатарзалните кости (обикновено скъсяване на четвъртия и по-рядко втория пръст) - така нареченият брахи-метафалангизъм. Отбелязва се калцификация на меките тъкани и подкожни калцификации, които могат да бъдат открити при раждането; често се наблюдава едновременно засягане на други ендокринни жлези: щитовидна жлеза (хипофункция), гонади, панкреас (захарен диабет). В резултат на хипокалцемия често се развиват катаракта и дефекти на зъбния емайл. Като диференциално диагностичен тест за разграничаване на PHP тип 1A от хипопаратироидизъм: липсата на клиничен ефект от парентералното приложение на ПТХ под формата на повишаване на нивото на калций в кръвта и увеличаване на бъбречната екскреция на фосфор в урината (фосфатен ефект).
Биохимичното изследване разкрива хипокалцемия, хиперфосфатемия, повишени нива на паратироидния хормон в кръвта и хипофосфатурия. Нивото на Gs протеин в кръвта е намалено. При рентгеново изследванеСкелетната система разкрива скъсяване на метакарпалните и метатарзалните кости, генерализирана деминерализация и удебеляване на костите на черепния свод.
ПСЕВДОХИПОПАРАТРОЗА ТИП 1Б
има автозомно доминантен тип наследяване, но не е изключен доминантен, Х-свързан тип наследяване. Необходимо е да се има предвид наблюдаваното понякога непълно проникване на гена на заболяването и възможността за латентно носителство на патологията. Поради това се препоръчва клинично (откриване на субклинично протичане на заболяването) и биохимично изследване (определяне на нивата на калций, фосфор, ПТХ в кръвта) на предполагаеми носители на заболяването. PHP тип 1B се причинява от дефицит на тъканни рецептори за паратиреоиден хормон в прицелните органи и ограничена резистентност към паратиреоиден хормон. Клиничната картина е подобна на тази при тип 1А, но няма увреждане на други ендокринни жлези, по-рядко се среща остеодистрофия.
Пациентите нямат бъбречен отговор на екзогенно приложение на паратиреоиден хормон под формата на повишена екскреция на цикличен аденозин монофосфат в урината; обаче, за разлика от тип 1А, нивото на Gs протеин в кръвта е нормално. Жените са засегнати по-често от мъжете, но тежестта на заболяването може да бъде еднаква както при мъжете, така и при жените.
ПСЕВДОХИПОПАРАТРОЗА ТИП 1C
някои автори го идентифицират с псевдо-псевдохипопаратироидизъм (PPHP), описан от Albright F. през 1952 г. Характеризира се с клинична картина, характерна за PHP, но нивата на калций и фосфор в кръвта и урината остават в нормални граници. Нивата на PTH и Gs протеин в кръвта също остават непроменени за нормално ниво. Някои пациенти с PHP тип 1C имат делеции de novoна хромозома 2. Възможно е този вариант на заболяването да е подвид на PGP тип 1А.
ПСЕВДОХИПОПАРАТРОЗА ТИП 2
клинично подобен на други видове заболяване, но има автозомно-рецесивен начин на наследяване. Не може да се изключи наличието на автозомно-доминантни форми на патология. Патогенезата на развитието е свързана с вътреклетъчна резистентност към сАМР. След това PTH се свързва с рецепторите и предизвиква нормален клетъчен отговор към PTH под формата на повишена екскреция на cAMP. Вътреклетъчната нечувствителност към сАМР обаче не позволява да се реализира пълният ефект на ПТХ. В същото време остава нормална реакциябъбреците до екзогенно приложение на паратироиден хормон под формата на повишена екскреция на цикличен аденозин монофосфат в урината. Предполага се, че PHP тип 2 може да бъде свързан с дефицит на витамин D.
По този начин идентифицираните видове PGP се характеризират клинично с намалена чувствителност на целевите органи към ПТХ, но се различават по патогенетичните механизми на образуване на тъканна нечувствителност.
ДИАГНОСТИКА
Лабораторен диференциално диагностичен тест може да бъде моделът на бъбречна екскреция на сАМР в отговор на прилагане на ПТХ: повишена екскреция на сАМР се наблюдава при тип 2 и липсата му при тип 1. Диагнозата се потвърждава от откриването на намалено ниво на гуанин нуклеотид свързващ протеин (Gs протеин) в кръвта (средно 1,5-2 пъти) в сравнение с нормата. Хипокалциемията обикновено се комбинира с хиперфосфатемия и хипофосфатурия. Нивата на ПТХ са повишени; при тип 1C нивото на ПТХ е нормално, което води до името „псевдохипопаратироидизъм“. Рентгеновото изследване на скелетната система разкрива скъсяване на метакарпалните и метатарзалните кости, често генерализирана деминерализация (остеопороза) и удебеляване на костите на черепния свод. Дерматоглифният модел показва изместване на аксиалния палмарен трирадиус.
Критерии за диагностика:
- нисък ръст;
- кръгло лице;
- нервно забавяне умствено развитие;
- скелетни аномалии;
- нисък серумен калций;
- високо ниво на паратхормон в кръвта;
- намалена уринна екскреция на фосфати и сАМР.
ЛЕЧЕНИЕ И ПРОФИЛАКТИКА
Лечението на хипокалцемия се състои в предписване на калциеви добавки в дози, достатъчни за поддържане на нормални концентрации на калций в кръвта. Голямо значение има терапията с витамин D. Понастоящем се използват активни метаболити на витамин D - оксидевит, 1-алфа-D3, калцитрин и др., в доза 1-2 mcg/ден с положителен резултат(повишени нива на калций в кръвта, намалени симптоми конвулсивен синдром). Тахистин (0,5–1,5 mg/ден) също е ефективен. Това лекарствоповишава абсорбцията на калций в червата и по този начин спомага за повишаване на нивото на калций в кръвта. Антиконвулсивната терапия се използва като допълнителна терапия. На интелектуално развитиелечението няма забележим ефект, но заедно с намаляването на симптомите на конвулсивен синдром се наблюдава регресия неврологични прояви(подкорови нарушения, хореиформна хиперкинеза, атетоза и др.). За да се избегне предозиране на препарати с витамин D, е необходимо да се следи концентрацията на калций в кръвта на всеки 3-7 дни през първите 2 седмици от лечението и всеки месец през следващите 2-3 месеца. След достигане на стабилна концентрация на калций в кръвта е достатъчно да се проверява веднъж на всеки 2-3 месеца. Диетата с ограничен прием на фосфор спомага за нормализиране на концентрациите на фосфор и калций в кръвта и премахва симптомите на вторичен хиперпаратироидизъм. При недостатъчност на други ендокринни жлези се провежда заместителна терапия с подходящи хормони.
Лечението с паратироиден хормон е неефективно. За облекчаване на конвулсивни атаки се прилага интравенозно 10% разтвор на калциев хлорид или калциев глюконат; перорално - 5-10% разтвор на калциев хлорид, 1 супена лъжица 3-4 пъти на ден: калциев глюконат, калциев лактат - до 10 g на ден.
ПРОГНОЗАза цял живот се определя от тежестта на конвулсивния синдром.
ПРЕДОТВРАТЯВАНЕзаболяване се основава на данни от медицинско генетично консултиране.
МЕДИКО-ГЕНЕТИЧНО КОНСУЛТИРАНЕ
При провеждане на медицинско генетично консултиране трябва да се изхожда от автозомно-доминантния тип наследяване и високия (50%) риск от повторение на заболяването в семейството с наследствени форми. За да се идентифицира естеството на вида на наследството, е необходимо да се извърши задълбочен преглед на родителите, тъй като синдромът може да се прояви минимално клинични симптоми. Понастоящем е разработена и усъвършенствана молекулярно-генетична диагностика на заболяването чрез типизиране на мутации в гена GNAS1 на хромозома 20. Разработват се методи за пренатална диагностика на заболяването като цяло и неговите отделни видове.
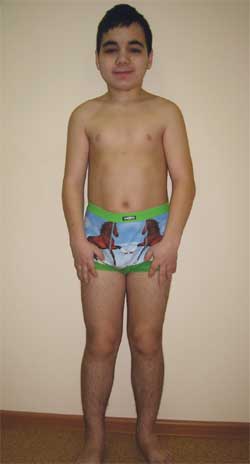
КЛИНИЧНО НАБЛЮДЕНИЕМомче Г., 14,5 години (снимка 4), е прието в Изследователския клиничен институт по педиатрия с диагноза: дегенеративно заболяване нервна система? вродена външна хидроцефалия; симптоматична епилепсия; наследствен синдром? болест на съхранението? метаболитна енцефалопатия; субклиничен хипотиреоидизъм; нисък ръст от смесен произход; когнитивно увреждане.
Оплакванияпри приемане на интензивни пароксизмални главоболия, локализирани в челната област и придружени от повръщане, което носи облекчение, намалена памет и ефективност в училище, конвулсивни атаки, по време на които се появяват потрепвания в дясната ръка.
Снимка 4.
Дете Г., 14,5 години, с остеодистрофия на Олбрайт
(фенотипни характеристики, нисък ръст, скъсени крайници, брахидактилия)
Семейна история:родителите са арменци по националност, нямат кръвна връзка и нямат професионални рискове. В родословието на случаите психично заболяване, епилепсия, забавяне на развитието не са отбелязани. Sibs, сестра, на 17 години, се казва, че е здрава.
История на живота и болестта:момче от 2-ра бременност, протекла без особености, второ раждане, на термин, физиологично, тегло - 3100 гр., дължина - 51 см. Извика веднага, оценка по Апгар - 7/9 точки. Влошаване на състоянието на 3-ия ден - неонатални гърчове, спрени в родилния дом. Ранният постнатален период е без особености. Имаше леко забавяне в двигателното развитие през първата година от живота, независимо ходене от 1 година 3 месеца. Във връзка с това той е наблюдаван от невролог с диагноза: органична лезияЦНС; вродена хидроцефалия; неонатални гърчове; анамнеза за фебрилни гърчове.
Получих диакарб и финлепсин. Начало на атаките на 1 година и 11 месеца. – асиметричен, тонизиращ под формата на напрежение дясна ръкаи краката, с отваряне на очите, до 2 минути, без загуба на съзнание, често до 10 епизода на ден. Получавах Депакин нередовно. На фона на независимо оттегляне - единичен тонизиращ статус. На 2-годишна възраст е извършена компютърна томография на мозъка по местоживеене, където са идентифицирани единични огнища на демиелинизация в тилните дялове.
Направена е консултация с неврохирург и е препоръчано консервативно лечение. От 3-годишна възраст се наблюдава изоставане в психо-речевото развитие и се препоръчва наблюдение от психиатър.
От 4-5-годишна възраст родителите започват да забелязват деформация и скъсяване на пръстите на ръцете и краката, особено на 2-4 пръста симетрично на ръцете и краката, както и намаляване на показателите за растеж. На 8-годишна възраст заключението на логопеда е общо нарушение на речта от 2-3-то ниво, препоръчва се обучение в специализирано училище. На същата възраст преглед от генетик по местоживеене, заключение: наследствено заболяванеобмен? Препоръчва се изследване на кръвните аминокиселини, не са открити промени; крайно заключение: не са установени данни за наследствено метаболитно заболяване; хипохондроплазия; Препоръчва се лечение при невролог и ендокринолог.
Таблица.
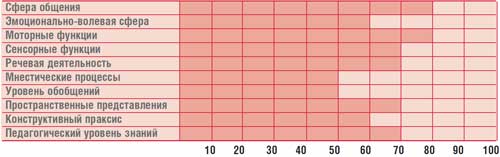
Профил на умственото развитие на дете Г., 14,5 години (IQ = 68)
На 8 години е консултиран от ендокринолог за забавен растеж и развитие. При рентгеново изследване на ръцете се установяват следните особености: средните и основните фаланги и метакарпалните кости са скъсени и удебелени; Диагнозата на рентгенолога е ахондроплазия.
Многократно прегледан по местоживеене в неврологична болница. На 12-годишна възраст се появиха конвулсивни пристъпи без загуба на съзнание с потрепвания на дясната ръка, които бяха серийни, беше назначена антиконвулсивна терапия (депакин), честотата на пристъпите значително намаля. На 13 години е направен ЯМР на мозъка с контраст - симетрични промени в основата на темпоралните дялове на ниво ядра под формата на повишаване на МР сигнала, което е характерно за токсични (манган) или метаболитни (мед, желязо) енцефалопатии.
Прегледана на 13 години 3 месеца. ендокринолог, изследването на профила на щитовидната жлеза разкрива увеличение тироид-стимулиращ хормон(TSH), диагностициран е субклиничен хипотиреоидизъм, предписан е L-тироксин.
При анализа на амбулаторната карта и документацията на детето по местоживеене е проведено изследване на калций и фосфор веднъж, на възраст от 1,5 години, е отбелязана хипокалциемия, но не е извършено допълнително изследване по този повод. Като се има предвид несигурността на диагнозата по местоживеене, генетикът изпрати детето в Москва, в Научноизследователския клиничен институт по педиатрия, за да изясни диагнозата.
Данни обективно изследване:
Височина – 143 см, тегло – 43 кг.
Физическото развитие е много ниско, хармонично, телосложението е непропорционално поради скъсяване на крайниците. Растежът Sds съответства на –2,8 отклонения от нормата (норма –2+2).
Фенотипни характеристики: кръгло лице, къса шия, антимонголоидни палпебрални фисури, широк мост на носа, високо чело, брахидактилия, скъсяване на IV и V метакарпални и метатарзални кости (снимка 5). Вътрешни органи – без особености. Сексуално развитие – етап III–IV по Танер (което съответства на възрастта).
Лаборатория и функционални изследвания:
Клиничен анализкръвта и урината са нормални.
Биохимичен анализкръв: общ калций– 1,39 (норма 2,02–2,6 mmol/l), йонизиран калций – 0,61 (норма 1,13–1,32 mmol/l), неорганичен фосфор – 3,66 (норма 0,86– 1,56 mmol/l), останалите показатели са в нормални граници.
Биохимичен анализ на урината: бъбречната екскреция на фосфати е намалена - 11,5 mmol/l (норма 19–32 mmol/l).
Профил на щитовидната жлеза: TSH – 11,75 (нормален диапазон 0,4–4,0 µIU/ml), свободен Т4 – 0,49 (нормален диапазон 1,0–1,8 ng/dL).
Паратироиден хормон – 499 (норма 12–65 pg/ml), STH – 7 ng/ml (норма 7–10 ng/ml), соматомедин-С – 250 ng/ml (норма 88–360 ng/ml).
Ултразвук вътрешни органи- без функции.
ЕКГ - миграция на суправентрикуларния пейсмейкър на фона на нормална сърдечна честота от 71-80 удара / мин. Непълна блокада на десния клон на пакета. Нарушаване на процеса на реполяризация в миокарда задна стеналява камера (намален z.T III, aVF).
R-графия на гръбначния стълб – дясностранна сколиоза гръднигръбначен стълб 1-ва степен, тежка остеопороза.
R-графия на ръцете с улавяне на предмишниците - скъсяване и разширяване на крайните и средните фаланги. Костна възраст – 13,5–14 години.
Не са регистрирани ЕЕГ модели на епилептична активност.
ЯМР на мозъка - ЯМР картина на множество субкортикални огнища на повишен МР сигнал във фронталните лобове, външна компенсирана хидроцефалия с атрофия на мозъчното вещество.
MSCT на мозъка показва симетрични области на калцификация на лентиформени ядра. Дифузни хиперденсни зони в таламуса, опашните ядра с област на калцификация вдясно. Множество точкови калцификации на покривните меки тъкани на черепа.
Аудиограма – без патология.
В ход е ДНК диагностика в гена GNAS1.
Специализирани консултации:
Ендокринолог – Наследствена остеодистрофия на Олбрайт тип 1А (псевдохипопаратироидизъм). Първичен хипотиреоидизъм, непълно обезщетение за лекарства.
Офталмолог – пълна вторична катаракта. Препоръчва се хирургично лечение.
Психолог – когнитивно увреждане (психологичният профил на детето е представен в таблицата).
Като се вземат предвид фенотипа на детето, медицинската история, резултатите допълнителни изследвания(хипокалциемия, хиперфосфатемия, хипофосфатурия, повишен паратиреоиден хормон в кръвта), калцификати в мозъка, наличие на катаракта, хипотиреоидизъм), поставена е диагноза: Наследствена остеодистрофия на Олбрайт тип 1А (псевдохипопаратироидизъм). Препоръчва се провеждането на ДНК диагностика - търсене на мутации в гена GNAS1.
Лечение:Детето се препоръчва да приема еутирокс в доза 100 mcg/ден; активен метаболит на витамин D - алфа-D3 (Teva) в доза 2 мкг/ден; калций (Sandoz) 2000 mg/ден; постоянно прилагане на антиконвулсивна терапия - финлепсин 800 mg/ден под наблюдението на невролог-епилептолог; занимания с логопед и психолог; енерготропна терапия (Elkar и коензим Q10 в дози, свързани с възрастта). Проследяване на показателите на фосфорно-калциевия метаболизъм, нивата на паратироидния хормон.
По този начин,Представеното клинично наблюдение показва трудностите на диференциално-диагностичното търсене, важността на навременното изследване на прости биохимични параметри (в случай на епилепсия е задължителен повторен скрининг на показателите на фосфорно-калциевия метаболизъм), резултатите от късната диагностика на генетично обусловено заболяване , необходимостта от интегриране на отделни признаци в общия фенотип на определено заболяване патологично състояниеза целенасочена, навременна диагностика на отделните форми наследствени заболявания. Навременната диагностика и изясняване на генезиса на всеки синдром са особено важни, тъй като ни позволяват да намерим оптималния подход за лечение на тези състояния и профилактика. възможни усложнения(до увреждане на детето); предотвратяване на повторна поява на наследствени заболявания в засегнатите семейства (медико-генетично консултиране). Това налага необходимостта лекарите от различни специалности ясно да се ориентират в потока на наследствено обусловената патология. Списъкът с литература е в редакцията.