
Функционална биохимия. Порфирия - причини, класификация на порфирията биохимия
Видове порфирии - Лекция, раздел Химия, ЛЕКЦИОНЕН КУРС ПО ОБЩА БИОХИМИЯ Остра интермитентна порфирия (ОИП) – Причина – Дефект.
Острата интермитентна порфирия (AIP) се причинява от дефект в гена, кодиращ PBG - deaminase.
Унаследява се по автозомно-доминантен начин. Има натрупване на ранни прекурсори на синтеза на хема: 5-ALA (5-ALA) и порфобилиноген (PBG).
Вродена еритропоетична порфирия - рядко вродено заболяване, унаследява се по автозомно-рецесивен начин. Характеризира се с ниска активност на уропорфириноген-III косинтазата и висока активност на уропорфириноген-I синтаза. Образуването на уропорфириноген-I значително надвишава синтеза на уропорфириноген-III (нормалният изомер в пътя на синтеза на хема). Въпреки че генетичното заболяване засяга всички клетки, то се появява предимно в еритропоетична тъкан по неизвестна причина. Пациентите отделят големи количества уропорфириноген-I и копропорфириноген-I в урината; които във въздуха спонтанно се окисляват до уропорфирин-I и копропорфирин-I – червени флуоресцентни пигменти. Циркулиращите еритроцити съдържат големи количества уропорфирин-I, но най-високата концентрация на този порфирин се наблюдава в клетките костен мозък.
Отбелязва се фоточувствителност на кожата поради активирането на LPO под въздействието на светлина върху порфириновите съединения. Пациентите имат пукнатини по кожата и често се наблюдават хемолитични явления.
Наследствената копропорфирия е автозомно доминантно заболяване, причинено от дефицит на копропорфириноген оксидаза, митохондриален ензим, отговорен за превръщането на копропорфириноген III в протопорфириноген IX. Копропорфириноген III c големи количестваах се отстранява от тялото с изпражнения и урина. Копропорфириногенът бързо се окислява на светлина и въздух, превръщайки се в червения пигмент копропорфирин.
Ограничената способност за синтезиране на хем (особено при стресови условия) води до дерепресия на ALA синтазата. В резултат на това се наблюдава прекомерно образуване на ALA, порфобилиноген и други междинни продукти на синтеза на хема. Пациентите проявяват всички признаци и симптоми, свързани с излишъка на ALA и порфобилиноген, които са характерни за интермитентна остра порфирия, и има повишена фоточувствителност поради наличието на излишни количества копропорфириногени и уропорфириногени.
Мозаечната порфирия или наследствената фоторопорфирия е автозомно доминантно заболяване, при което ензимното превръщане на протопорфириногена в хем е частично блокирано. Обикновено това превръщане се извършва от два ензима, протопорфириноген оксидаза и ферохелатаза, локализирани в митохондриите. Съдържанието на протопорфириноген оксидаза е само половината от нормалното количество. Пациентите с мозаечна порфирия проявяват относителен дефицит на съдържание на хем при стресови условия, както и дерепресирано състояние на чернодробната ALA синтаза. Което води до свръхпроизводство на всички междинни продукти на синтеза на хема в зоните преди блокирания стадий. Пациентите с мозаечна порфирия отделят излишни количества ALA, порфобилиноген, уропорфирин и копропорфирин в урината си и отделят уропорфирин, копропорфирин и протопорфирин в изпражненията. Урината на пациентите е пигментирана и флуоресцира, а кожата е чувствителна към светлина по същия начин, както при пациенти с porphyria cutanea tarda (виж по-долу).
Porphyria cutanea tarda е най-честата форма на порфирия. Обикновено се свързва с увреждане на черния дроб, особено с прекомерна консумация на алкохол или претоварване с железни йони. Вероятната причина е частичен дефицит на уропорфириноген декарбоксилаза. Разстройството изглежда се предава автозомно доминантна черта, но генетичната пенетрантност варира и в повечето случаи зависи от наличието на чернодробна дисфункция. Урината съдържа повишени количества уропорфирини тип I и III; в същото време уринарната екскреция на ALA и порфобилиноген се наблюдава относително рядко. Понякога урината съдържа много значително количество порфирини, което й придава розов оттенък; когато се подкисли, най-често произвежда розова флуоресценция в ултравиолетовата област.
Тази тема принадлежи към раздела:
ЛЕКЦИОНЕН КУРС ПО ОБЩА БИОХИМИЯ
Катедра по биохимия. ЛЕКЦИОНЕН КУРС ПО ОБЩА БИОХИМИЯ.
Ако се нуждаеш допълнителен материалпо тази тема или не сте намерили това, което търсите, препоръчваме да използвате търсенето в нашата база данни с произведения: Видове порфирии
Какво ще правим с получения материал:
Ако този материал е бил полезен за вас, можете да го запазите на страницата си в социалните мрежи:
Всички теми в този раздел:
Кръвта е течна тъкан, която изпълнява редица важни функции в организма: Основната функция на кръвта е транспортирането на вещества и топлинна енергия; а) Дихателна функция.
Всички телесни течности имат редица общи свойства: обем, плътност, вискозитет, pH и осмотично налягане. Нормални стойностиОбщи свойства на кръвта на възрастни:
Формените елементи (клетки) на кръвта съставляват 45% от общия кръвен обем. № Кръвни клетки Концентрация % от общия обем до
Химичният състав на веществата, разтворими в кръвната плазма, е относително постоянен, тъй като има мощни нервни и хуморални механизми, които поддържат хомеостазата.
Показател Възраст 1 ден 1 месец. 6 месеца 1 година 1-6 л 12 л
В кръвната плазма са открити повече от 200 вида протеини, които съставляват 7% от обема на плазмата. Протеините на кръвната плазма се синтезират главно в черния дроб и макрофагите, както и в съдовия ендотел, червата, лимфата
Структурата на протеините на кръвната плазма е глобуларна, според състава те се делят на прости (албумин) и сложни. Сред сложните могат да се разграничат липопротеините (VLDL, LPPP, LDL, HDL, CM), gly
Основният протеин на тази фракция е албуминът. албумин. Прост протеин от 585 АА с маса 69 kDa, има 17 дисулфидни моста, много дикарбоксилни АА, има висока
VLDL – образуват се в черния дроб. Транспорт TG, HS. DILI – образува се в кръвта от VLDL. Транспорт TG, HS.
Те се синтезират от функционално активни В-лимфоцити (плазмоцити). Един възрастен има 107 клонинга на В-лимфоцити, които синтезират 107 вида γ-глобулини. γ-глобула
Концепцията за „протеини на острата фаза“ обединява до 30 протеина на кръвната плазма, участващи във възпалителния отговор на организма към увреждане. Протеините на острата фаза се синтезират в черния дроб, техният край
Ензимите, намиращи се в кръвната плазма, могат да се разделят на 3 основни групи: 1. Секреторни. Те се синтезират в черния дроб, чревния ендотел, навлизат в кръвоносните съдове.
Факултети: терапевтични и превантивни, медицински и превантивни, педиатрични. 2-ри курс. Червените кръвни клетки (erythrosytus) са профилирани елементикръв,
Червените кръвни клетки при хора и бозайници в кръвния поток обикновено (80%) имат формата на двойновдлъбнати дискове и се наричат дискоцити. Тази форма на червени кръвни клетки създава най-голямата площ
Олигозахаридите (сиалова киселина и антигенни олигозахариди) на гликолипидите и гликопротеините, разположени на външната повърхност на плазмената мембрана, образуват гликокаликса.
В зрял еритроцит протеините не се синтезират, т.к няма рибозоми, ER, апарат на Голджи или ядро. Пептидът глутатион обаче се синтезира в цитоплазмата. Биосинтезата на глутатион протича в
В зрял еритроцит: 1. AMP може да се синтезира от PRPP (от рибоза-5ph) и аденин. 2. АМФ с участието на АТФ се превръща в АДФ. 3. При реакции на субстратно фосфорилиране
През деня до 3% от хемоглобина може спонтанно да се окисли до метхемоглобин: Hb (Fe2+) ® Met Hb (Fe3+) +e- Редукция на метхемоглобина до хемоглобин
Хемът е порфирин, в центъра на който има
Хемът е простатна група от много протеини: хемоглобин, миоглобин, митохондриални CPE цитохроми, цитохром Р450, каталазни ензими, пероксидаза, цитохромоксидаза, триптофан
Хемът се синтезира във всички тъкани, но най-често в костния мозък и черния дроб. В костния мозък хемът е необходим за синтеза на хемоглобин, в хепатоцитите - за образуването на цитохром Р450
Хем, синтезиран в митохондриите, индуцира синтеза на глобинови вериги върху полирибозомите. Гените на глобиновата верига са разположени на хромозоми 11 и 16. Глобиновите вериги образуват глобули и се свързват с хе
Хемоглобинът е тетрамерен хромопротеин, има маса 64,5 kDa, състои се от 4 хема и 4 глобина. Глобините са представени от полипептидни вериги различни видове a, b, g, d и т.н. верига
Хемоглобинът със свободна шеста желязна координационна връзка в хема се нарича апохемоглобин. Шестата координационна връзка може да свързва различни лигани
Хемоглобинът свързва O2 последователно, по една молекула за всеки хем. В апохемоглобина, поради координационната връзка с протеиновата част, атома
Сътрудничеството в работата на протомерите на хемоглобина формира сигмоидния характер на кривата на неговото насищане с кислород в зависимост от парциалното налягане на кислорода. S-образна крива на насищане
Ефектът на рН върху естеството на кривата на дисоциация на оксихемоглобина се нарича ефект на Бор (на името на датския физиолог Кристиан Бор, който пръв открива този ефект). &nb
2,3-DPG намалява афинитета на хемоглобина към кислорода и по този начин увеличава доставката на кислород до тъканите. Ако кръвта е изразходвала целия си запас от DPG, хемоглобинът остава практически наситен с кислород
1) ембрионални хемоглобини (Gover I, Gover II). В ранните етапи на развитие на плода през първите седмици от развитието, когато се появяват огнища на хематопоеза в жълтъчната торбичка
Анормалните хемоглобини са резултат от мутации в гените, кодиращи a и b веригите. Известни са няколкостотин мутантни човешки хемоглобини (в повечето случаи функционално активни).
Заболяванията на хемоглобините се наричат хемоглобинози, те са повече от 200. Хемоглобинозите се делят на хемоглобинопатии и таласемии. Хемоглобинопатии
Старите увредени червени кръвни клетки се фагоцитират от RES клетки и се усвояват в лизозомите. При разпадането на хемоглобина се образува жлъчният пигмент билирубин. Допълнителен катаболизъм на билирубина в черния дроб, киш
Искате ли да получавате последните новини по имейл?
Абонирайте се за нашия бюлетин
Новини и информация за студенти
реклама
Свързан материал
- Подобен
- Популярен
- Облак от етикети
- Тук
- Временно
- празна
Относно сайта
Информацията под формата на резюмета, записки, лекции, курсови работи и дисертации има свой автор, който притежава правата. Ето защо, преди да използвате каквато и да е информация от този сайт, уверете се, че не нарушавате нечии права.
Биохимия на порфирия
Хемът, желязосъдържащо тетрахидропиролно багрило, е компонент на O 2 -свързващи протеини (виж стр. 274) и различни оксидоредуктазни коензими (виж стр. 108, 310). Почти 85% от биосинтезата на хема се извършва в костния мозък и само малка част в черния дроб. Митохондриите и цитоплазмата участват в синтеза на хема.
Синтезът на тетрахидропиролови пръстени започва в митохондриите. От сукцинил-КоА (схема по-горе), междинен продукт в цитратния цикъл, кондензацията с глицин произвежда продукт, чието декарбоксилиране произвежда 5-аминолевулинат (ALA). Отговорна за този етап, 5-аминолевулинат синтазата (ALA синтаза) [1] е ключовият ензим на целия път. Експресията на синтеза на ALA синтаза се инхибира от хема, т.е. крайния продукт и съществуващия ензим. Това е типичен случай на инхибиране на крайния продукт или инхибиране с обратна връзка.
След синтеза 5-аминолевулинатът преминава от митохондриите в цитоплазмата, където две молекули се кондензират в порфобилиноген, който вече съдържа пиролов пръстен [2]. Порфобилиноген синтазата [2] се инхибира от оловни йони. Следователно, когато остро отравянеолово се намира в кръвта и урината повишени концентрации 5-аминолевулинат.
На следващите етапи се образува тетрапиролна структура, характерна за порфирина. Свързването на четири порфобилиногенови молекули с елиминирането на NH2 групите и образуването на уропорфириноген III се катализира от хидроксиметилбилан синтаза [3]. Образуването на този междинен продукт изисква втори ензим, уропорфириноген III синтаза [4]. Липсата на този ензим води до образуването на "грешен" изомер - уропорфириноген I.
Тетрапиролната структура на уропорфириноген III все още е значително различна от хема. Така централният железен атом липсва и пръстенът съдържа само 8 вместо 11 двойни връзки. В допълнение, пръстените носят само заредени R странични вериги (4 ацетатни и 4 пропионатни остатъка). Тъй като хем групите в протеините функционират в неполярна среда, е необходимо полярните странични вериги да бъдат преобразувани в по-малко полярни. Първо, четири ацетатни остатъка (R 1) се декарбоксилират до образуване на метилови групи (5). Полученият копропорфириноген III се връща в митохондриите. По-нататъшните стъпки се катализират от ензими, които са локализирани върху/или вътре в митохондриалната мембрана. Първо, под действието на оксидазата две пропионатни групи (R 2) се превръщат във винилови групи (6). Модификацията на страничните вериги завършва с образуването на протопорфириноген IX.
На следващия етап, поради окисление, в молекулата се създава спрегната π-електронна система, която придава характерния червен цвят на хема. Това изразходва 6 редуциращи еквивалента (7). Накрая, с помощта на специален ензим, ферохелатаза, двувалентен железен атом се включва в молекулата (8). Така образуваният хем или Fe-протопорфирин IX е включен например в хемоглобина и миоглобина (вижте стр. 274), където е свързан нековалентно, или в цитохром С, към който е свързан ковалентно (вижте стр. 108). ).
Има редица заболявания, причинени от наследствени или придобити нарушения на синтеза на порфирини, така наречените порфирии; някои от тях са много трудни. Много от тези заболявания водят до освобождаване на прекурсори на хема в изпражненията или урината, които могат да бъдат тъмночервени на цвят. Наблюдава се и отлагане на порфирини в кожата. При излагане на светлина това причинява мехури, които трудно зарастват. При порфириите също се среща често неврологични разстройства. Възможно е в основата на средновековните легенди за човешки вампири (Дракули) да е странното поведение на пациенти с порфирия (фотофобия, необичаен външен вид и поведение, консумация на кръв, която компенсира недостига на хем и често подобрява състоянието при някои форми на порфирия ).
Хемоглобинът има молекулярни заболявания
Сърповидно-клетъчна анемия
HbS – сърповидноклетъчен хемоглобин. При това нарушение в ДНК в резултат на точкова мутация триплетът CTT се заменя с триплет CAT, което води до включването на аминокиселината валин в 6-та позиция на β-веригата вместо глутамат. Промяната в свойствата на β-веригата води до промяна в свойствата на цялата молекула и образуването на "лепкава" област на повърхността на хемоглобина. Когато хемоглобинът е деоксигениран, мястото се "отваря" и свързва една молекула деоксихемоглобин S с други подобни. Резултатът е полимеризация на молекулите на хемоглобина и образуване на големи протеинови нишки, причиняващи деформация на еритроцита и хемолиза при преминаване през капилярите.
Схема на разликата между хемоглобин S и хемоглобин А и неговата полимеризация
Нарушен синтез на хемоглобин
Порфирия
Порфириите са група от разнородни наследствени заболявания, в резултат на нарушен синтез на хем и повишено съдържание на порфирини и техните прекурсори в организма. Има наследствени и придобити форми на порфирия.
Придобитите форми на порфирия са токсични по природа и се причиняват от действието на хексахлоробензен, оловни соли и други тежки метали (инхибиране на порфобилиноген синтаза, ферохелатаза и др.), Лекарства (противогъбичен антибиотик гризеофулфин).
При наследствените форми ензимният дефект присъства във всички клетки на тялото, но се проявява само в един тип клетки. Има два големи групипорфирия:
1. Чернодробни - група заболявания с автозомно-доминантни нарушения на ензимите на различни етапи от синтеза на протопорфирин IX. Най-яркото заболяване от тази група е интермитентната остра порфирия, при която при хетерозиготите активността на уропорфириноген I синтазата е намалена с 50%.
Заболяването се проявява след достигане на пубертета поради повишената нужда на хепатоцитите от цитохром Р 450 за неутрализиране на половите стероиди; обостряне на състоянието често се случва след приемане на лекарства, чийто метаболизъм изисква участието на цитохром Р 450. Намаляването на концентрацията на хема, използван за синтеза на цитохром Р 450, активира аминолевулинат синтазата. В резултат на това пациентите отделят големи количества порфобилиноген и аминолевулинова киселина в урината. На светлина порфириногенът се окислява до оцветени порфобилин и порфирин, което е причината за потъмняването на урината, когато тя стои на светлина с достъп на въздух. Симптомите включват остра коремна болка, запек, сърдечно-съдови нарушения и невропсихиатрични разстройства.
2. Еритропоетични - автозомно-рецесивни нарушения на определени ензими за синтеза на протопорфирин IX в еритроидните клетки. В този случай балансът на реакциите при образуването на уропорфириногени се измества към синтеза на уропорфириноген I. Симптомите на заболяването са подобни на предишния, но освен това има фоточувствителност на кожата поради наличието на уропорфириногени, в освен това има пукнатини по кожата и хемолитични явления.
Таласемия
Таласемията се характеризира с намаляване на синтеза на α-вериги на хемоглобина (α-таласемия) или β-вериги (β-таласемия). Това води до нарушена еритропоеза, хемолиза и тежка анемия.
Можете да попитате или да оставите вашето мнение.
Видове порфирии
Нарушения на синтеза на хема. Порфирия
Порфириите са хетерогенна група от заболявания, причинени от нарушения в синтеза на хема, дължащи се на дефицит на един или повече ензими.
Няма единна класификация на порфириите. Порфириите се разделят по причини на:
1) Наследствена. Възниква, когато има дефект в гена за ензима, участващ в синтеза на хема;
2) Закупени. Те възникват поради инхибиторния ефект на токсични съединения (хексохлоробензен, соли на тежки метали - олово) върху ензимите на синтеза на хема.
В зависимост от преобладаващата локализация на ензимния дефицит (в черния дроб или червените кръвни клетки), порфиринът се разделя на:
1) чернодробна– най-често срещаният тип порфирин включва остра интермитентна порфирия (AIP), porphyria cutanea tarda, наследствена копропорфирия, мозаечна порфирия;
2) еритропоетичен– вродена еритропоетична порфирия (болест на Гюнтер), еритропоетична протопорфирия.
Зависи от клинична картина, порфириите се разделят на:
Отрицателните последици от порфирията са свързани с дефицит на хема и натрупване в тъканите и кръвта на междинни продукти от синтеза на хема - порфириногени и продукти на тяхното окисление. При еритропоетични порфирии порфирините се натрупват в нормобластите и еритроцитите, при чернодробните порфирии - в хепатоцитите.
За всеки тип порфирия има определено ниво на ензимен дефект, в резултат на което се натрупват продукти, синтезирани над това ниво. Тези продукти са основните диагностични маркери на заболяването.
Порфириногените са отровни тежки формипорфирии те причиняват невропсихиатрични разстройства, дисфункция на RES и увреждане на кожата.
Невропсихиатричните разстройства при порфириите са свързани с факта, че аминолевулинатите и порфириногените са невротоксини.
В кожата на слънце порфириногените лесно се превръщат в порфирини. Кислородът, когато взаимодейства с порфирините, преминава в синглетно състояние. Синглетният кислород стимулира LPO на клетъчните мембрани и разрушаването на клетките, така че порфириите често са придружени от фоточувствителност и язви на откритите кожни участъци.
Порфириногените не са оцветени или флуоресцират, но порфирините проявяват интензивна червена флуоресценция в ултравиолетовите лъчи. Излишните порфирини, които се отделят с урината, го дават тъмен цвят(„порфирин“ означава лилаво на гръцки).
Понякога при леки форми на наследствена порфирия заболяването може да бъде асимптоматично, но приемането на лекарства, които са индуктори на синтеза на аминолевулинатна синтаза, може да доведе до обостряне на заболяването. В някои случаи симптомите на заболяването не се появяват до пубертета, когато повишеното производство на β-стероиди предизвиква индукция на синтеза на аминолевулинатна синтаза. Порфирия се наблюдава и при отравяне с оловни соли, тъй като оловото инхибира аминолевулинат дехидратазата и ферохелатазата. Някои халоген-съдържащи хербициди и инсектициди са индуктори на синтеза на аминолевулинатна синтаза, така че тяхното поглъщане е придружено от симптоми на порфирия.
Острата интермитентна порфирия (AIP) се причинява от дефект в гена, кодиращ PBG - deaminase. Унаследява се по автозомно-доминантен начин. Има натрупване на ранни прекурсори на синтеза на хема: 5-ALA (5-ALA) и порфобилиноген (PBG).
Безцветният PBG на светлина се превръща в порфибилин и порфирин, те придават тъмен цвят на урината. ALA има невротоксичен ефект, водещ до вяла парализа на крайниците и пареза на дихателната мускулатура. Последното причинява остър дихателна недостатъчност. Заболяването се проявява в средна възраст и се провокира от приема на аналгетици и сулфатични лекарства, тъй като те повишават синтеза на ALA синтаза.
Клиничните симптоми включват остра коремна болка, повръщане, запек, сърдечно-съдови нарушения и нервно-психични разстройства. Невидим свръхчувствителностна светлина, тъй като метаболитното разстройство възниква на етапа, предшестващ образуването на уропорфириноген.
За лечение се използва лекарството нормозанг - хем аргинат. Действието се основава на факта, че хемът, чрез механизъм за отрицателна обратна връзка, блокира транслацията на ALA синтазата и следователно синтезът на ALA и PBG намалява, което постига облекчаване на симптомите.
Вродената еритропоетична порфирия е още по-рядко вродено заболяване, унаследено по автозомно-рецесивен начин. Молекулярната природа на това заболяване не е точно известна; установено е обаче, че се характеризира с известен дисбаланс в относителните активности на уропорфириноген III-косинтазата и уропорфириноген-1 синтазата. Образуването на уропорфириноген I е количествено значително по-добро от синтеза на уропорфириноген III - нормален изомер в пътя на синтеза на хема. Въпреки че генетичното заболяване засяга всички клетки, то се появява предимно в еритропоетична тъкан по неизвестна причина. Пациентите с вродена еритропоетична порфирия отделят големи количества от тип I изомери на уропорфириноген и копропорфириноген; в урината и двете съединения се окисляват спонтанно в уропорфирин I и копропорфирин I, червени флуоресцентни пигменти. Съобщава се за случай, при който се наблюдава леко повишаване на концентрацията на уропорфирин III, но съотношението на тип I към III изомери е приблизително 100:1. Циркулиращите еритроцити съдържат големи количества уропорфирин 1, но най-високата концентрация на този порфирин се наблюдава в клетките на костния мозък (но не и в хепатоцитите).
Има светлинна чувствителност на кожата поради естеството на абсорбционния спектър на порфириновите съединения, които се образуват в големи количества. Пациентите имат пукнатини по кожата и често се наблюдават хемолитични явления.
Наследствената копропорфирия е автозомно доминантно заболяване, причинено от дефицит на копропорфириноген оксидаза, митохондриален ензим, отговорен за превръщането на копропорфириноген III в протопорфириноген IX. Копропорфириноген III се отстранява от тялото в големи количества с изпражненията и също така, поради разтворимостта си във вода, се екскретира в големи количествас урина. Подобно на уропорфириногена, копропорфириногенът бързо се окислява на светлина и въздух, превръщайки се в червения пигмент копропорфирин.
Ограничената способност за синтез на хем при това заболяване (особено при стресови условия) води до дерепресия на ALA-цитазата. В резултат на това има прекомерно образуване на ALA и порфобилиноген, както и други междинни продукти по пътя на синтеза, образувани на етапи, предхождащи наследствено блокирания стадий. Съответно пациентите с наследствена копропорфирия проявяват всички признаци и симптоми, свързани с излишъка на ALA и порфобилиноген, които са характерни за интермитентна остра порфирия, но в допълнение те имат повишена фоточувствителност поради наличието на излишни количества копропорфириногени и уропорфириногени. При това заболяване прилагането на хематин може също така да причини поне частично потискане на ALA синтазата и да облекчи симптомите, дължащи се на свръхпроизводството на хем биосинтетични междинни продукти.
Мозаечната порфирия или наследствената фоторопорфирия е автозомно доминантно заболяване, при което ензимното превръщане на протопорфириногена в хем е частично блокирано. Обикновено това превръщане се извършва от два ензима, протопорфириноген оксидаза и ферохелатаза, локализирани в митохондриите. Съдейки по данните, получени върху култивирани кожни фибробласти, при пациенти с мозаечна порфирия съдържанието на протопорфириноген оксидаза е само половината от нормалното количество. Пациентите с мозаечна порфирия проявяват относителен дефицит на съдържание на хем при стресови условия, както и дерепресирано състояние на чернодробната ALA синтаза. Както беше отбелязано по-горе, повишената активност на ALA синтазата води до свръхпроизводство на всички междинни съединения на синтеза на хема в местата нагоре по веригата на блокирания етап. Така пациентите с мозаечна порфирия отделят излишни количества ALA, порфобилиноген, уропорфирин и копропорфирин в урината и отделят уропорфирин, копропорфирин и протопорфирин в изпражненията. Урината на пациентите е пигментирана и флуоресцира, а кожата е чувствителна към светлина по същия начин, както при пациенти с porphyria cutanea tarda (виж по-долу).
Porphyria cutanea tarda е може би най-честата форма на порфирия. Обикновено се свързва с някакъв вид увреждане на черния дроб, особено с прекомерна консумация на алкохол или претоварване с железни йони. Естеството на метаболитното нарушение не е точно установено, но вероятна причинае частичен дефицит на уропорфириноген декарбоксилаза. Разстройството изглежда се предава като автозомно доминантна черта, но генетичната пенетрантност варира и в повечето случаи зависи от наличието на чернодробна дисфункция. В съответствие с прогнозите, урината съдържа повишени количества уропорфирини тип I и III; в същото време уринарната екскреция на ALA и порфобилиноген се наблюдава относително рядко. Понякога урината съдържа много значително количество порфирини, което й придава розов оттенък; когато се подкисли, най-често произвежда розова флуоресценция в ултравиолетовата област.
Черният дроб съдържа големи количества порфирини и следователно флуоресцира силно, докато червените кръвни клетки и клетките на костния мозък не флуоресцират. Основната клинична проява на porphyria cutanea tarda е повишената фоточувствителност на кожата. Пациентите не изпитват повишена активност на ALA синтазата или, съответно, излишни нива на порфобилиноген и ALA в урината; това корелира с липсата на остри пристъпи, характерни за интермитентна остра порфирия.
Протопорфирията или еритропоетичната протопорфирия изглежда се причинява от доминантно наследствен дефицит на ферохелатазна активност в митохондриите на всички тъкани; Клинично това заболяване се проявява като остра уртикария, причинена от излагане на слънчева светлина. Червените кръвни клетки, плазмата и изпражненията съдържат повишени количества протопорфирин IX, а ретикулоцитите (незрели червени кръвни клетки) и кожата (когато се изследват чрез биопсия) често флуоресцират в червено. Черният дроб вероятно също допринася за повишеното образуване на протопорфирин IX, но не се наблюдава екскреция на порфирини и техните прекурсори в урината.
Биохимия на порфирия
Порфириите са хетерогенна група от заболявания, характеризиращи се с повишена секреция на порфирини или техни прекурсори. Някои форми на порфирия са наследствени, други са придобити. Предложени са няколко различни класификациипорфирия. Удобно е да се разделят наследствените форми на три големи групи - еритропоетични, чернодробни и тези форми, при които метаболитните нарушения се наблюдават едновременно в еритропоетични и чернодробни тъкани (Таблица 33.2). Повечето наследствени форми се характеризират с наличие на метаболитен
Таблица 33.2. Класификация на човешките порфирии
физически нарушения във всички тъкани, но по някаква причина се появяват в един вид тъкан. По-долу е дадено описание на биохимичните нарушения, характерни за порфириите.
Всеки тип порфирия се характеризира с набор от порфирини и техните прекурсори, екскретирани в урината. Тези данни и тяхната връзка с различните етапи на синтеза на хема са показани на фиг. 33.11.
Интермитентната остра пневмония (IAP) е характеристика на автозомно доминантно заболяване наследствено заболяванепри хората, което обикновено се появява едва след достигане на пубертета. Причината за него е наследствен частичен дефицит на уропорфириноген-1 синтаза. Пациентите са хетерозиготни за дефектен структурен ген, така че активността на уропорфириноген-1 синтазата в техните клетки е 50% от нормалната. Пациентите с ПОП отделят големи количества порфобилиноген и ALA в урината. И двете съединения са безцветни, но порфобилиногенът спонтанно образува два цветни продукта на светлина и въздух - порфобилин и порфирин. Това е причината урината да потъмнява, когато стои на светло с достъп на въздух.
Порфобилиногенът и ALA присъстват в плазмата и цереброспиналната течност на пациентите, особено по време на остри екзацербации. Лекарствата и стероидните хормони, чийто метаболизъм изисква участието на хем-съдържащи протеини, като цитохром Р-450, могат да ускорят появата на обостряне. Съединенията, които индуцират порфирия по време на метаболизма, увеличават консумацията на хем протеини и по този начин намаляват вътреклетъчната концентрация на хема; това води до дерепресия на синтеза на ALA синтаза. Повишената активност на ALA синтазата и частичното блокиране на уропорфириноген-1 синтазата води до значително натрупване на ALA и порфобилиноген; това е придружено от остри болкив корема, повръщане, запек, сърдечно-съдови нарушения, както и нервно-психични разстройства. Трябва да се отбележи, че според експерименталните данни намаляването на съдържанието на хем инхибира активността на триптофан пиролазата и води до натрупване на невроактивни съединения - триптофан и 5-хидрокситриптамин.
Пациентите с ПОП не изпитват повишената чувствителност към светлина, която е характерна за други чернодробни порфирии. Това можеше да се очаква, тъй като нито порфирините, нито порфириногените се натрупват в пациентите, тъй като метаболитното разрушаване на синтеза на хема настъпва на етапа, предшестващ образуването на първия порфириноген (уропорфириноген).
Както беше отбелязано по-горе, метаболитни нарушения се откриват и в други клетки, по-специално в червените кръвни клетки, както и в култивирани фибробласти или клетки от амниотична течност; въпреки това повишената активност на ALA синтазата, водеща до свръхпроизводство на ALA и порфобилиноген, се проявява предимно в черния дроб. Очевидно това се дължи на факта, че черният дроб е самият орган, в който се извършва метаболизмът на индуциращите агенти. Острата порфирия е един от редките примери за заболяване, което е фенотипно изразено в хетерозиготи, докато ензимният дефицит е само 50%.
Както може да се предвиди въз основа на
Ориз. 33.11. Последователни етапи на биосинтезата на хема, показващи прекурсорите, екскретирани в урината по време на различни формипорфирия Къдравите скоби комбинират съединения, които се екскретират в урината в излишни количества по време на обостряне на тези форми на порфирия. ALA - 5-аминолевулинова киселина. (Възпроизведено с модификации от статията Kaufman L., Merver N. S. Biochemical defects in two types of human hepatic prphyria. N. Engl. J. Med 1970:283:954.)
предложеният механизъм за регулиране на синтеза на ALA синтаза от репресивната система - дерепресин, прилагането на хематин при пациенти с POP може да намали индукцията на ALA синтаза и по този начин да облекчи хода на заболяването.
Вродената ертропоетична порфирия е още по-рядко вродено заболяване, унаследено по автозомно-рецесивен начин. Молекулярната природа на това заболяване не е точно известна; установено е обаче, че се характеризира с известен дисбаланс в относителните активности на уропорфириноген-III косинтазата и уропорфириноген-1 синтазата. Образуването на уропорфириноген I е количествено значително по-добро от синтеза на уропорфириноген III, нормален изомер в пътя на синтеза на хема. Въпреки че генетичното заболяване засяга всички клетки, то се появява предимно в еритропоетична тъкан по неизвестна причина. Пациентите с вродена еритропоетична порфирия отделят големи количества от тип I изомери на уропорфириноген и копропорфириноген; в урината и двете съединения се окисляват спонтанно до уропорфирин 1 и копропорфирин I - червени флуоресцентни пигменти. Съобщава се за случай, при който се наблюдава леко повишаване на концентрацията на уропорфирин III, но съотношението на тип I към III изомери е приблизително 100:1. Циркулиращите еритроцити обаче съдържат големи количества уропорфирин I най-висока концентрацияТози порфирин се наблюдава в клетките на костния мозък (но не и в хепатоцитите).
Вероятно поради образуването на по-малки количества от истинския прекурсор на хема, уропорфириноген III, и произтичащия относителен дефицит на хема в еритропоетичните тъкани на пациентите, се индуцира ALA синтаза. Тази индукция води до свръхпроизводство на порфириногени тип I. Заедно с повишения синтез на ALA синтаза и свръхпроизводството на порфириногени тип I, образуването и екскрецията на порфобилиноген и ALA се увеличава. По този начин, въз основа на биохимични аномалии, е възможно да се предвиди появата клинични симптоми, подобни на наблюдаваните при POP, но в допълнение има фоточувствителност на кожата,
поради естеството на абсорбционния спектър на порфириновите съединения, които се образуват в големи количества. Пациентите имат пукнатини по кожата и често се наблюдават хемолитични явления.
Наследствената автозомна копропорфирия е доминантно заболяване, причинено от дефицит на копропорфириноген окндаза, митохондриален ензим, отговорен за превръщането на копропорфириноген III в протопорфириноген IX. Копропорфириноген III се отстранява от тялото в големи количества с изпражненията, а също така, поради разтворимостта си във вода, се екскретира в големи количества с урината. Подобно на уропорфириногена, копропорфириногенът бързо се окислява на светлина и въздух, превръщайки се в червения пигмент копропорфирин.
Ограничената способност за синтез на хем при това заболяване (особено при стресови условия) води до дерепресия на ALA синтазата. В резултат на това има прекомерно образуване на ALA и порфобилиноген, както и други междинни продукти по пътя на синтеза на хема, образувани на етапи, предхождащи наследствено блокирания стадий. Съответно пациентите с наследствена копропорфирия проявяват всички признаци и симптоми, свързани с излишъка на ALA и порфобилиноген, които са характерни за интермитентна остра порфирия, но в допълнение те имат повишена фоточувствителност поради наличието на излишни количества копропорфириногени и уропорфириногени. При това заболяване прилагането на хематин може също така да причини поне частично потискане на ALA синтазата и да облекчи симптомите, дължащи се на свръхпроизводството на хем биосинтетични междинни продукти.
Мозаечната порфирия или наследствената фотокопропорфирия е автозомно доминантно заболяване, при което ензимното превръщане на протопорфириногена в хем е частично блокирано. Обикновено това превръщане се извършва от два ензима, протопорфириноген оксидаза и ферохелатаза, локализирани в митохондриите. Съдейки по данните, получени върху култивирани кожни фибробласти, при пациенти с мозаечна порфирия съдържанието на протопорфириноген оксидаза е само половината от нормалното количество. Пациентите с мозаечна порфирия проявяват относителен дефицит на съдържание на хем при стресови условия, както и дерепресирано състояние на чернодробната ALA синтаза. Както беше отбелязано по-горе, повишената активност на ALA синтазата води до свръхпроизводство на всички междинни съединения на синтеза на хема в местата нагоре по веригата на блокирания етап. Така пациентите с мозаечна порфирия отделят излишни количества ALA, порфобилиноген, уропорфирин и копропорфирин в урината и отделят уропорфирин, копропорфирин и протопорфирин в изпражненията. Урината на пациентите е пигментирана и флуоресцира, а кожата е чувствителна към светлина по същия начин, както при пациенти с porphyria cutanea tarda (виж по-долу).
Porphyria cutanea tarda е може би най-честата форма на порфирия. Обикновено се свързва с някакъв вид увреждане на черния дроб, особено с прекомерна консумация на алкохол или претоварване с железни йони. Естеството на метаболитното нарушение не е точно установено, но вероятната причина е частичен дефицит на уропорфрин декарбоксилаза. Разстройството изглежда се предава като автозомно доминантна черта, но генетичната пенетрантност варира и в повечето случаи зависи от наличието на чернодробна дисфункция. В съответствие с прогнозите, урината съдържа повишени количества уропорфирини тип I и III; в същото време уринарната екскреция на ALA и порфобилиноген се наблюдава относително рядко. Понякога урината съдържа много значително количество порфирини, което й придава розов оттенък; когато се подкисли, най-често произвежда розова флуоресценция в ултравиолетовата област.
Черният дроб съдържа големи количества порфирини и следователно флуоресцира силно, докато червените кръвни клетки и клетките на костния мозък не флуоресцират. Основен клинична изяваПри porphyria cutanea tarda има повишена фоточувствителност на кожата. Пациентите не изпитват повишена активност на ALA синтазата или, съответно, излишни нива на порфобилиноген и ALA в урината; това корелира с липсата на остри пристъпи, характерни за интермитентна остра порфирия.
Протопорфирията или еритропоетичната протопорфирия изглежда се причинява от доминантно наследствен дефицит на ферохелатазна активност в митохондриите на всички тъкани; Клинично това заболяване се проявява като остра уртикария, причинена от излагане на слънчева светлина. Червените кръвни клетки, плазмата и изпражненията съдържат повишени количества протопорфирин IX, а ретикулоцитите (незрели червени кръвни клетки) и кожата (когато се изследват чрез биопсия) често флуоресцират в червено.
Черният дроб вероятно също допринася за повишеното образуване на протопорфирин IX, но не се наблюдава екскреция на порфирини и техните прекурсори в урината.
Придобитата (токсична) пневмония може да бъде причинена от действието на токсични съединения като хексахлоробензен, оловни соли и други тежки метали, както и лекарства като гризеофулвин. Тежки металиса инхибитори на редица ензими в системата за синтез на хем, включително ALA дехидратаза, уропорфириноген синтаза и ферохелатаза.
Раздел I. Структура и функция на белтъци и ензими
Раздел II. Биоенергетика и метаболизъм на въглехидрати и липиди
Раздел III. Метаболизъм на протеини и аминокиселини
Копирането на информация от страницата е разрешено само с връзка към този сайт
Порфириите са хетерогенна група от заболявания, характеризиращи се с повишена секреция на порфирини или техни прекурсори. Някои форми на порфирия са наследствени, други са придобити. Предложени са няколко различни класификации на порфириите. Удобно е да се разделят наследствените форми на три големи групи - еритропоетични, чернодробни и тези форми, при които метаболитните нарушения се наблюдават едновременно в еритропоетични и чернодробни тъкани (Таблица 33.2). Повечето наследствени форми се характеризират с наличие на метаболитен
Таблица 33.2. Класификация на човешките порфирии
физически нарушения във всички тъкани, но по някаква причина се появяват в един вид тъкан. По-долу е дадено описание на биохимичните нарушения, характерни за порфириите.
Всеки тип порфирия се характеризира с набор от порфирини и техните прекурсори, екскретирани в урината. Тези данни и тяхната връзка с различните етапи на синтеза на хема са показани на фиг. 33.11.
Интермитентната остра порфирия (IAP) е характеристика на автозомно доминантно наследствено заболяване при човека, което обикновено не се проявява до след пубертета. Причината за него е наследствен частичен дефицит на уропорфириноген-1 синтаза. Пациентите са хетерозиготни за дефектен структурен ген, така че активността на уропорфириноген-1 синтазата в техните клетки е 50% от нормалната. Пациентите с ПОП отделят големи количества порфобилиноген и ALA в урината. И двете съединения са безцветни, но порфобилиногенът спонтанно образува два цветни продукта на светлина и въздух - порфобилин и порфирин. Това е причината урината да потъмнява, когато стои на светло с достъп на въздух.
Порфобилиногенът и ALA присъстват в плазмата и цереброспиналната течност на пациентите, особено по време на остри екзацербации. Лекарствата и стероидните хормони, чийто метаболизъм изисква участието на хем-съдържащи протеини, като цитохром Р-450, могат да ускорят появата на обостряне. Съединенията, които индуцират порфирия по време на метаболизма, увеличават консумацията на хем протеини и по този начин намаляват вътреклетъчната концентрация на хема; това води до дерепресия на синтеза на ALA синтаза. Повишената активност на ALA синтазата и частичното блокиране на уропорфириноген-1 синтазата води до значително натрупване на ALA и порфобилиноген; това е придружено от остра коремна болка, повръщане, запек, сърдечно-съдови нарушения, както и нервно-психични разстройства. Трябва да се отбележи, че според експерименталните данни намаляването на съдържанието на хем инхибира активността на триптофан пиролазата и води до натрупване на невроактивни съединения - триптофан и 5-хидрокситриптамин.
Пациентите с ПОП не изпитват повишената чувствителност към светлина, която е характерна за други чернодробни порфирии. Това можеше да се очаква, тъй като нито порфирините, нито порфириногените се натрупват в пациентите, тъй като метаболитното разрушаване на синтеза на хема настъпва на етапа, предшестващ образуването на първия порфириноген (уропорфириноген).
Както беше отбелязано по-горе, метаболитни нарушения се откриват и в други клетки, по-специално в червените кръвни клетки, както и в култивирани фибробласти или клетки от амниотична течност; въпреки това повишената активност на ALA синтазата, водеща до свръхпроизводство на ALA и порфобилиноген, се проявява предимно в черния дроб. Очевидно това се дължи на факта, че черният дроб е самият орган, в който се извършва метаболизмът на индуциращите агенти. Острата порфирия е един от редките примери за заболяване, което е фенотипно изразено в хетерозиготи, докато ензимният дефицит е само 50%.
Както може да се предвиди въз основа на

Ориз. 33.11. Последователни етапи на биосинтеза на хема, показващи прекурсорите, екскретирани в урината при различни форми на порфирия. Къдравите скоби комбинират съединения, които се екскретират в урината в излишни количества по време на обостряне на тези форми на порфирия. ALA - 5-аминолевулинова киселина. (Възпроизведено с модификации от статията Kaufman L., Merver N. S. Biochemical defects in two types of human hepatic prphyria. N. Engl. J. Med 1970:283:954.)
предложеният механизъм за регулиране на синтеза на ALA синтаза от репресивната система - дерепресин, прилагането на хематин при пациенти с POP може да намали индукцията на ALA синтаза и по този начин да облекчи хода на заболяването.
Вродената ертропоетична порфирия е още по-рядко вродено заболяване, унаследено по автозомно-рецесивен начин. Молекулярната природа на това заболяване не е точно известна; установено е обаче, че се характеризира с известен дисбаланс в относителните активности на уропорфириноген-III косинтазата и уропорфириноген-1 синтазата. Образуването на уропорфириноген I е количествено значително по-добро от синтеза на уропорфириноген III, нормален изомер в пътя на синтеза на хема. Въпреки че генетичното заболяване засяга всички клетки, то се появява предимно в еритропоетична тъкан по неизвестна причина. Пациентите с вродена еритропоетична порфирия отделят големи количества от тип I изомери на уропорфириноген и копропорфириноген; в урината и двете съединения се окисляват спонтанно до уропорфирин 1 и копропорфирин I - червени флуоресцентни пигменти. Съобщава се за случай, при който се наблюдава леко повишаване на концентрацията на уропорфирин III, но съотношението на тип I към III изомери е приблизително 100:1. Циркулиращите еритроцити съдържат големи количества уропорфирин I, но най-висока концентрация на този порфирин се наблюдава в клетките на костния мозък (но не и в хепатоцитите).
Вероятно поради образуването на по-малки количества от истинския прекурсор на хема, уропорфириноген III, и произтичащия относителен дефицит на хема в еритропоетичните тъкани на пациентите, се индуцира ALA синтаза. Тази индукция води до свръхпроизводство на порфириногени тип I. Заедно с повишения синтез на ALA синтаза и свръхпроизводството на порфириногени тип I, образуването и екскрецията на порфобилиноген и ALA се увеличава. По този начин, въз основа на биохимични аномалии, е възможно да се предвиди появата на клинични симптоми, подобни на тези, наблюдавани при POP, но в допълнение се отбелязва фоточувствителност на кожата,
поради естеството на абсорбционния спектър на порфириновите съединения, които се образуват в големи количества. Пациентите имат пукнатини по кожата и често се наблюдават хемолитични явления.
Наследствената автозомна копропорфирия е доминантно заболяване, причинено от дефицит на копропорфириноген окндаза, митохондриален ензим, отговорен за превръщането на копропорфириноген III в протопорфириноген IX. Копропорфириноген III се отстранява от тялото в големи количества с изпражненията, а също така, поради разтворимостта си във вода, се екскретира в големи количества с урината. Подобно на уропорфириногена, копропорфириногенът бързо се окислява на светлина и въздух, превръщайки се в червения пигмент копропорфирин.
Ограничената способност за синтез на хем при това заболяване (особено при стресови условия) води до дерепресия на ALA синтазата. В резултат на това има прекомерно образуване на ALA и порфобилиноген, както и други междинни продукти по пътя на синтеза на хема, образувани на етапи, предхождащи наследствено блокирания стадий. Съответно пациентите с наследствена копропорфирия проявяват всички признаци и симптоми, свързани с излишъка на ALA и порфобилиноген, които са характерни за интермитентна остра порфирия, но в допълнение те имат повишена фоточувствителност поради наличието на излишни количества копропорфириногени и уропорфириногени. При това заболяване прилагането на хематин може също така да причини поне частично потискане на ALA синтазата и да облекчи симптомите, дължащи се на свръхпроизводството на хем биосинтетични междинни продукти.
Мозаечната порфирия или наследствената фотокопропорфирия е автозомно доминантно заболяване, при което ензимното превръщане на протопорфириногена в хем е частично блокирано. Обикновено това превръщане се извършва от два ензима, протопорфириноген оксидаза и ферохелатаза, локализирани в митохондриите. Съдейки по данните, получени върху култивирани кожни фибробласти, при пациенти с мозаечна порфирия съдържанието на протопорфириноген оксидаза е само половината от нормалното количество. Пациентите с мозаечна порфирия проявяват относителен дефицит на съдържание на хем при стресови условия, както и дерепресирано състояние на чернодробната ALA синтаза. Както беше отбелязано по-горе, повишената активност на ALA синтазата води до свръхпроизводство на всички междинни съединения на синтеза на хема в местата нагоре по веригата на блокирания етап. Така пациентите с мозаечна порфирия отделят излишни количества ALA, порфобилиноген, уропорфирин и копропорфирин в урината и отделят уропорфирин, копропорфирин и протопорфирин в изпражненията. Урината на пациентите е пигментирана и флуоресцира, а кожата е чувствителна към светлина по същия начин, както при пациенти с porphyria cutanea tarda (виж по-долу).
Porphyria cutanea tarda е може би най-честата форма на порфирия. Обикновено се свързва с някакъв вид увреждане на черния дроб, особено с прекомерна консумация на алкохол или претоварване с железни йони. Естеството на метаболитното нарушение не е точно установено, но вероятната причина е частичен дефицит на уропорфрин декарбоксилаза. Разстройството изглежда се предава като автозомно доминантна черта, но генетичната пенетрантност варира и в повечето случаи зависи от наличието на чернодробна дисфункция. В съответствие с прогнозите, урината съдържа повишени количества уропорфирини тип I и III; в същото време уринарната екскреция на ALA и порфобилиноген се наблюдава относително рядко. Понякога урината съдържа много значително количество порфирини, което й придава розов оттенък; когато се подкисли, най-често произвежда розова флуоресценция в ултравиолетовата област.
Черният дроб съдържа големи количества порфирини и следователно флуоресцира силно, докато червените кръвни клетки и клетките на костния мозък не флуоресцират. Основната клинична проява на porphyria cutanea tarda е повишената фоточувствителност на кожата. Пациентите не изпитват повишена активност на ALA синтазата или, съответно, излишни нива на порфобилиноген и ALA в урината; това корелира с липсата на остри пристъпи, характерни за интермитентна остра порфирия.
Протопорфирията или еритропоетичната протопорфирия изглежда се причинява от доминантно наследствен дефицит на ферохелатазна активност в митохондриите на всички тъкани; Клинично това заболяване се проявява като остра уртикария, причинена от излагане на слънчева светлина. Червените кръвни клетки, плазмата и изпражненията съдържат повишени количества протопорфирин IX, а ретикулоцитите (незрели червени кръвни клетки) и кожата (когато се изследват чрез биопсия) често флуоресцират в червено.
Черният дроб вероятно също допринася за повишеното образуване на протопорфирин IX, но не се наблюдава екскреция на порфирини и техните прекурсори в урината.
Придобитата (токсична) пневмония може да бъде причинена от действието на токсични съединения като хексахлоробензен, оловни соли и други тежки метали, както и лекарства като гризеофулвин. Тежките метали са инхибитори на редица ензими в системата за синтез на хема, включително ALA дехидратаза, уропорфириноген синтаза и ферохелатаза.
Урс А. Майер (Урс А. Майер)
Порфириите са патологии, свързани с наследствени или придобити аномалии в биосинтезата на хема. Порфирините, тези тетрапиролни пигменти, действат като междинни продукти в този път и се образуват от прекурсори - 8 -аминолевулинова киселина (ALA) и порфобилиноген. Хем, комплекс от двувалентно желязо с протопорфирин IX, функционира като простетична група от хемопротеини като хемоглобин, цитохроми, каталаза и триптофан оксигеназа. Неговата биосинтеза е жизненоважна и се случва във всички аеробни клетки.
Всяка порфирия се характеризира с характеристики на хиперпродукция, натрупване и екскреция на междинни продукти от биосинтезата на хема. Тези характеристики отразяват метаболитната експресия на дефицита на отделни биосинтетични ензими.
Основните клинични прояви са периодични епизоди на дисфункция нервна системаи/или чувствителност на кожата към слънчева светлина. Неврологичният синдром обикновено се предизвиква от лекарства като барбитурати и се състои от коремна болка, периферна невропатия и психични разстройства. Невропсихичните симптоми се появяват само при порфирии, при които рязко се увеличава производството на порфиринови прекурсори - ALA и порфобилиноген. Патогенезата на неврологичните заболявания е неясна. Фоточувствителността на кожата е пряко свързана с повишеното натрупване на порфирини, въпреки че различни нарушениякожните прояви са различни. Фоточувствителността се дължи на фотодинамичния ефект на порфирините и вероятно се медиира от образуването на синглетен кислород с последващо развитие на деструктивни процеси, като липидна пероксидация на лизозомни мембрани. Доминантно унаследените човешки порфирии се изразяват различно. Могат да се определят само биохимични или ензимни промени. Подобен латентен курсЗаболяването може да бъде един от етапите или да продължи през целия живот на пациента. В други случаи симптомите могат да бъдат причинени от лекарства, хормони или увреждане на черния дроб.
Класификация. Порфириите обикновено се разделят на две основни групи (еритропоетични и чернодробни) в съответствие с основните места на синтеза на хема, в които се появяват метаболитни "грешки". Единствената чисто еритропоетична форма на порфирия, вродената еритропоетична (CEP), е рядка. При протопорфирията (PrP) порфирините се натрупват както в еритроидните клетки, така и в чернодробната тъкан. При интермитентна остра порфирия, наследствена копропорфирия и разноцветна порфирия (съответно IOP, NCP и PP), доминантно наследственият ензимен дефицит причинява нарушаване на биосинтезата на хема главно в черния дроб без видими нарушения в образуването на хемоглобин. Хроничната кожна порфирия (CCP) преди това се считаше за придобита чернодробна порфирия. Въпреки това, повечето (ако не всички) пациенти имат наследствен дефицит на уропорфириноген декарбоксилаза. Придобитата порфирия, наподобяваща CCP, се причинява от излагане на полихлорирани въглеводороди и чернодробни тумори. Отравянето с олово също е придружено от нарушен синтез на порфирини и хем. Известно повишение на уринарната екскреция на порфирини или техните прекурсори, както и натрупване на порфирини в еритроцитите, може да придружава много клинични състояния. При вторични явления няма симптоми или признаци на порфирия.
Биохимични аспекти. Последователността от реакции за синтеза на хем от субстратите глицин и сукцинил ензим А чрез ALA и порфобилиноген (PBA) се катализира от четири митохондриални и четири цитозолни ензима. Има разлики в регулацията на биосинтезата на хема в различните тъкани.
В черния дроб скоростта на образуване на хем е ограничена от реакцията, катализирана от ALA синтаза. Ензимите, действащи след ALA синтазата, се откриват в излишък. Основният регулатор на ALA синтазата е крайният продукт на целия път - хем, който потиска ензима чрез механизъм на отрицателна обратна връзка. Повишените нужди от хем се задоволяват чрез образуването на ALA синтаза. Неговият синтез в черния дроб се индуцира от голям брой мастноразтворими вещества, стероиди и химични съединения, които служат като субстрати и индуктори на хемопротеинови цитохроми Р 450 - крайните оксидази по пътя на микрозомалния метаболизъм на фармакологичните агенти. Тази индукция се модулира от множество генетични, метаболитни и заобикаляща среда. При порфирии, при които симптомите се провокират от определени лекарства, взаимозависимостта на синтеза на хема и микрозомалното окисление на тези лекарства става от голямо значение.
В клетките на костния мозък, където се осъществява пълен синтез на хема, ограничаващата скоростта реакция също се катализира от ALA синтазата, но малко се знае за нейната роля в синтеза на хема по време на деленето, диференциацията и узряването на еритроидните клетки. По време на узряването на тези клетки ядрата и митохондриите и следователно митохондриалните ензими изчезват от тяхсинтеза на хема, докато цитозолните ензими, които катализират реакцията между ALK и копро-порфириногена, се запазват. В това отношение червените кръвни клетки могат да се използват за диагностициране на порфирии, свързани с дефект само на цитозолен ензим.
Регулацията на синтеза на хема в костния мозък и черния дроб е различна. В черния дроб основният определящ фактор за образуването на хем е нивото на ALA синтазата, докато в костния мозък синтезът на хем се задейства от сложния процес на диференциация на еритроидни клетки. Ето защо вероятно дефектите в ензимите за синтез на хем в еритроидните клетки и черния дроб се проявяват по различен начин.
Порфириногените заемат междинна позиция между порфобилиноген и протопорфирин. Те са безцветни и не флуоресцират. С изключение на протопорфирина, порфирините са странични продукти, които напускат биосинтетичния път поради необратимо окисление на съответния порфириноген. Порфирините не изпълняват физиологична функция, но поради своя цвят и флуоресценция определят необичаен цвятурина и червени кръвни клетки при някои пациенти.
Местоположението на двете заместени странични вериги върху пироловия пръстен на порфирините определя структурните типове изомери, които са номерирани от I до IV. В природата се срещат само типове I и III, като само тип III служи като субстрат за крайните етапи на реакцията, водеща до образуването на протопорфирин IX и хем. Когато хемът се разпада, не се образуват порфирини, а нециклични тетрапироли, наречени жлъчни пигменти.
Вродена еритропоетична порфирия
Определение.Вродената еритропоетична порфирия (CEP, болест на Gunther, вродена фоточувствителна порфирия, еритропоетична уропорфирия) е рядко, рецесивно наследствено заболяване, проявяващо се като хронична фоточувствителност с обезобразяващи кожни лезии и хемолитична анемия.
Генетика, честота и патогенеза.Пациентите са хомозиготи за автозомно рецесивен ген. При хетерозиготите метаболизмът на порфирин рядко се нарушава, външно те изглеждат здрави. Основният ензимен дефект не е установен, но може да е функционален дисбаланс в активността на порфобилиноген деаминазата и уропорфириноген косинтазата III . Тази аномалия се изразява изключително в зреещи еритроидни клетки и води до рязка хиперпродукция на уропорфириногеназ , докато производството на уропорфириноген III не се променя или леко се увеличава. Уропорфириногеназ не може да се използва за синтез на хем, но се превръща в копропорфириногеназ Уропорфирин I , копропорфириногеназ и копропорфириназ натрупват се в тъканите и се отделят в излишни количества с урината и изпражненията.
Клинични прояви и диагноза.При пациентите порфирините се натрупват по време на развитието на плода. Още по време на или малко след раждането обикновено започва да се отделя розова или червена урина, докато фоточувствителност на кожата, периодична хемолиза и спленомегалия могат да се появят по-късно. Често се откриват хипертрихоза и червено оцветяване на зъбите и костите. Смъртта може да настъпи още в детството. При по-продължителна преживяемост пациентът развива големи, осакатяващи белези, особено по кожата на пръстите, носа и ушите. В урината се определят големи количества уропорфирин I, копропорфирин и порфирини със 7, 6, 5 и 3 карбоксилни групи, докато екскрецията на AL K и PBG не се променя. В изпражненията се откриват големи количества копропорфирин I. Нормобластите, ретикулоцитите и еритроцитите съдържат големи количества уропорфириназ и малко количество копропорфириногеназ . Нормобластите и ретикулоцитите проявяват интензивна червена флуоресценция. В съответствие с нормалната екскреция на ALK и PBG, няма неврологична патология.
Лечение. Излагането на слънчева светлина трябва да се избягва. В някои случаи хемолитичната анемия, екскрецията на порфирин и фоточувствителността намаляват след спленектомия. Използване на инфузии с хематин и перорално приложение Р-каротинът все още не е излязъл извън рамките на експеримента.
Чернодробни порфирии
Трите чернодробни порфирии (IOP, NCP и PP) са сходни по много начини. всичкоте се наследяват като автозомно доминантен белег. Острите пристъпи на животозастрашаваща неврологична патология се провокират от различни лекарства, хормони и други фактори, при които големи количества ALA и PBG се екскретират в урината, но видовете порфирини в урината и изпражненията са различни.
Интермитентна остра порфирия.Определение. Интермитентната остра порфирия [ВОН, остра интермитентна порфирия (AIP), пиролопорфирия] се характеризира с повтарящи се пристъпи на неврологични и психиатрични симптоми. Няма фоточувствителност. Основно е нарушена функцията на порфобилиноген деаминазата.
Генетика, честота и патогенеза. Аномалията се унаследява като автозомно-доминантен белег с променлива експресивност. Анормалният ген се среща с честота 1:10 000-1:50 000, но в някои региони може да е по-висока. Не са намерени хомозиготи. Причината за заболяването е частичен (50%) дефицит на порфобилиноген дезаминаза, която превръща PBG в уропорфириноген I. На генетично ниво този дефицит може да бъде причинен от повече от един механизъм, но най-честата мутация причинява намаляване на количеството на имунореактивния протеинов ензим. В черния дроб частичният ензимен дефицит води до повишена активност и/или индуцируемост на ALA синтазата лекарстваи други фактори и, следователно, до увеличаване на образуването и екскрецията на ALA и PBG в урината. При тези условия порфирините не се натрупват и следователно фоточувствителността на кожата не се повишава. При IOP се открива намалена активност на порфобилиноген деаминазата в черния дроб, еритроцитите, култивираните кожни фибробласти, левкоцитите и клетките на амниотичната течност. По този начин ензимният дефект се среща и в екстрахепаталните тъкани, но неговите метаболитни последици не се проявяват в тях. Ензимният дефицит при липса на придобити фактори не води непременно до клинично значима остра порфирия и само 1/3 пациенти и дори по-малко, страдащи от този генетичен дефект, някога са преживели пристъп на порфирия.Връзката между генетичния дефект и неврологичните разстройства остава неясно.
Клинични прояви и диагноза. Симптомите на заболяването се проявяват рядко до пубертет. Обикновено първият и най-очевиден симптом на пристъп на порфирия е коремна болка. Може да бъде умерено или много силно, с колики, локализирано или генерализирано, излъчващо се към гърба или долната част на гърба. Болката вероятно е свързана с автономна невропатия, придружена от нарушена двигателна активност на стомашно-чревния тракт с редуване на спазматични и разширени участъци на червата. Коремът обикновено е мек и болката не се засилва при натиск. Тъй като температурата и левкоцитозата често са свързани, остър пристъп на порфирия може да имитира всеки възпалителен процесв коремната кухина. Тежко повръщане и запек са чести. Неврологичните и психичните аномалии се проявяват по различни начини. Функциите на периферните нерви, автономната нервна система, мозъчния ствол и черепномозъчни нервиили мозък. Тахикардия и лабилна хипертония с постурална хипотония, задържане на урина и прекомерно изпотяване. Хипертонията и тахикардията корелират с повишената екскреция на катехоламини. Периферна невропатиясе причинява от участието на предимно моторни нерви в процеса, но може да се присъедини и сензорен компонент. Дълбоките сухожилни рефлекси са намалени или липсват. Характерни са невритни болки в крайниците, зони на хипо- и парестезии, както и отпуснато увисване на краката и ръцете. Може да се развие параплегия или пълна отпусната квадриплегия. В миналото водещата причина за смърт е била парализата на дихателната мускулатура. При ангажиране на черепномозъчните нерви в процеса може да атрофира зрителния нерв, да се появи офталмоплегия и дисфагия. При по-тежко увреждане на централната нервна система се появяват делириум, кома и конвулсии. Въпреки обратимостта на невропатията, остатъчната пареза може да персистира няколко години след остър пристъп. Много пациенти остават раздразнителни и емоционално нестабилни за дълго време с персистиращи функционални разстройства. При 1/3 от пациентите психиката е нарушена, може да се развие органичен мозъчен синдром с тревожност, дезориентация и зрителни халюцинации. Понякога се открива тежка хипонатриемия. Това може да се дължи на няколко причини (включително екскреция на натрий през стомашно-чревния тракт, ненужно приложение на течности и нефропатия с загуба на сол поради токсичния ефект на ALA), но водещата изглежда е неадекватната секреция на антидиуретичен хормон. В някои случаи хипомагнезиемията е толкова тежка, че се развива тетания.
Острите пристъпи продължават дни или дори месеци и варират по честота и тежест. По време на периоди на ремисия симптомите на заболяването отслабват или изчезват напълно. Клиничните (и биохимичните) прояви могат да бъдат провокирани от обичайни (терапевтични) дози барбитурати, антиконвулсанти, естрогени, контрацептиви или алкохол. Всички тези вещества се окисляват от хемопротеините на системата цитохром Р450. По време на остри пристъпи метаболизмът на някои от тях в черния дроб може да бъде нарушен. При някои жени влошаването на състоянието корелира с менструален цикъли латентна порфирия може да стане очевидна в късни датибременност или малко след раждането. Атаките могат да бъдат предизвикани и от дълъг период на намален прием на калории (гладуване).
Лабораторни данни. Острите пристъпи се характеризират с прекомерна екскреция с урината на ALA и PBG и в това отношение IOP не се различава от NCP или PP. Нивото на ALA и PBG в урината не корелира с тежестта на симптомите. Прост и надежден скринингов тест, който помага за диагностицирането на остра атака на IOP, NCP и PP, е качественото определяне на порфобилиноген в урината (тестове на Watson-Schwartz или Hosch). При невропсихиатрични разстройства тези тестове са почти винаги положителни, но изискват концентрациите на PBG в урината да бъдат 3 до 5 пъти над горната граница на нормата. В тази връзка и двата теста могат да бъдат отрицателни в случай на латентна форма на заболяването или нормализиране на екскрецията на PBG след спиране на атаката. Понякога се изисква количествено определяне ALA и PBG се екскретират в урината с помощта на хроматографски методи. При латентна форма на IOP с нормална екскреция на ALA и PBG, диагнозата може да се установи въз основа на резултатите от определяне на активността на порфобилиноген деаминазата в еритроцити, левкоцити или култивирани кожни фибробласти. Въпреки това, при здрави пациенти и пациенти с ВОН тези резултати се припокриват и точната диагноза не винаги е възможна.
При IOP, в съответствие с ензимния дефект, се увеличава екскрецията на порфиринови прекурсори - ALA и PBG, поради което прясно получената урина обикновено е безцветна и съдържа малко предварително образуван уро- или копропорфирин. Когато стои, може да потъмнее, тъй като PBG спонтанно се полимеризира в уропорфирин и порфобилин, тъмнокафяв пигмент с неизвестна структура. При някои пациенти обаче се определя достатъчно количество неензимно произведени пигменти, за да придаде тъмночервен цвят на прясно получената урина. Концентрацията на порфирини в изпражненията обикновено е в нормални граници.
Конвенционалните чернодробни функционални тестове са непроменени, с изключение на повишеното задържане на бромсулфалеин. В периферната кръв масата на еритроцитите е леко намалена, обемът на кръвта също намалява или се отбелязва преходна нормохромна нормоцитна анемия. Метаболитните промени по време на остър пристъп включват хиперхолестеролемия, придружена от повишени нива на липопротеини с ниска плътност, повишени серумни нива на тироксин (без хипертиреоидизъм), нарушен глюкозен толеранс и 5а-възстановяване на тестостерон в черния дроб. Връзката на тези аномалии с генетичен дефект остава неясна.
Лечение. Лечението по време на остър пристъп е еднакво за IOP, NCP и PP. Някои остри пристъпи изглежда се контролират чрез прилагане на големи количества (500 g/ден) въглехидрати (глюкоза ефект), въпреки че няма обективни проучвания за ефективността на това лечение. Препоръчва се венозно приложениеглюкоза със скорост 20 g/h. Ако състоянието на пациента не се подобри след 48 часа непрекъснато приложение на глюкоза или ако невропсихичните симптоми прогресират, трябва да се приложи хематин интравенозно (4 mg/kg за 10-15 минути на всеки 12 часа в продължение на 3-6 дни). Предлага се в търговската мрежа (панхематин) като лиофилизиран прах. Разтворите се приготвят непосредствено преди инфузията. Когато се използва хематин в препоръчителните дози, усложненията се развиват изключително рядко. Понякога се съобщава за тромбофлебит на мястото на инфузия, коагулопатия (проявяваща се с тромбоцитопения, удължаване на протромбиновото време, известна промяна в тромбопластиновото време и хипофибриногенемия) и хемолиза. Както хематинът, така и глюкозата при експериментални животни противодействат на индукцията на чернодробната ALA синтаза и в рамките на 48 часа могат да неутрализират биохимичните промени и да доведат до подобряване на състоянието на пациента. За предотвратяване и/или коригиране на хипонатриум, хипомагнезий и азотемия е важно да се проведе поддържащо лечение с внимателно проследяване на състоянието на водния и електролитния метаболизъм. Тахикардията и хипертонията трябва да се лекуват с бета-блокери. Ако диагнозата не се постави навреме, когато неврологичните симптоми прогресират, се свързват остри пристъпи висок рискна смъртта. Повечето пациенти се възстановяват напълно, но неврологичните симптоми могат да персистират месеци или години. Най-важно е да се предотврати остър пристъп, като се инструктира пациентът да избягва излагане на отключващи фактори като лекарства, стероиди, консумация на алкохол или умишлено гладуване.
Т.П. Харисън. Принципи на вътрешната медицина.Превод от доктор на медицинските науки А. В. Сучкова, д-р. Н. Н. Заваденко, д-р. Д. Г. Катковски
Заболявания, свързани с нарушен синтез на хем и често се проявяват с анемия, кожна сенсибилизация и различни неврологични разстройства. Един от първите случаи е описан от Шулц през 19 век – 1874г.
Идентифицирани различни видове порфирия, всеки от които е свързан с дефект в един от осемте ензима, участващи в синтеза на хема (с изключение на 5-аминолевулинат синтетазата). Установени са гените, кодиращи тези ензими и тяхната хромозомна локализация. Молекулярното увреждане, което е в основата на различните видове заболявания, е до голяма степен известно.
Схема на биосинтеза на хемаALA - 5-аминолевулинова киселина, PBG - порфобилиноген, UPG - уропорфириноген, AGE - копропорфириноген, PPG - протопорфириноген
Биосинтетичен блок, който възниква в резултат на ензимни дефекти, е най-силно изразен в черния дроб и костния мозък – органите, в които се синтезира основното количество хем. Всеки тип порфирия се характеризира с клинични и патоморфологични особености, отразяващи дефект в определен ензим и типа на унаследяване.
Като цяло за порфириясе характеризира с две осн клиничен синдром: кожна фоточувствителност и синдром на неврологично разстройство. Фотосенсибилизацията на кожата е резултат от реакцията на отложените в кожата порфирини към слънчевата радиация. Неврологичните разстройства се причиняват от повишено производство и екскреция на порфириновите прекурсори ALA и порфобилиноген. Когато два или повече ензима, участващи в синтеза на хема, са дефектни, се диагностицира двойна порфирия.
Генетични и метаболитни нарушения при порфирии Забележка. 1) * - процент от стойността нормална дейностензим; 2) основният метаболит и пътят на екскреция са подчертани с удебелен шрифт; 3) съкращения: ALA - 5-аминолевулинова киселина, PBG - порфобилиноген, UPG - уропорфириноген, CPG - копропорфириноген, PPG - протопорфириноген.
Забележка. 1) * - процент от стойността нормална дейностензим; 2) основният метаболит и пътят на екскреция са подчертани с удебелен шрифт; 3) съкращения: ALA - 5-аминолевулинова киселина, PBG - порфобилиноген, UPG - уропорфириноген, CPG - копропорфириноген, PPG - протопорфириноген.
Класификация на порфириите
аз Порфирии с кожна фоточувствителност:
- Вродена еритропоетична порфирия
- Porphyria cutanea tarda
- Протопорфирия
II. Остри или индуцирани порфирии:
- Порфирия с неврологични прояви
- Остра интермитентна порфирия
- ALK-D порфирия
- Порфирия с неврологични и кожни прояви
- Вариегатна порфирия
- Копропорфирия
III. Двойни порфирии
Хемът е простетична група от много протеини: хемоглобин, миоглобин, цитохроми на митохондриални CPE, цитохром Р 450, участващи в микрозомалното окисление. Ензимите каталаза, пероксидаза и цитохромоксидаза съдържат хем като коензим.
Всички клетки на тялото имат протеини, съдържащи хем, така че синтезът на хем се извършва във всички клетки, с изключение на еритроцитите, които, както е известно, нямат система за синтез на протеини.
Когато хемът се разпада в RPE клетките, се образува жлъчният пигмент билирубин. По-нататъшният катаболизъм на билирубина в черния дроб, червата и бъбреците води до образуването на крайни продукти на разпадане на хема стеркобилин и уробилин, съдържащи се съответно в изпражненията и урината. Желязото, отделено при разграждането на хема, отново се използва за синтеза на желязосъдържащи протеини.
I. СТРУКТУРА И БИОСИНТЕЗА НА HEM a. структура на хема
Хемът се състои от железен йон и порфирин (Фигура 13-1). Структурата на порфирините се основава на порфин. Порфинът се състои от четири пиролови пръстена, свързани с метенови мостове (фиг. 13-1). В зависимост от структурата на заместителите в пироловите пръстени се разграничават няколко вида порфирини: протопорфирини, етиопорфирини, мезопорфирини и копропорфирини. Протопорфирините са предшественици на всички останали видове порфирини.
Хемове от различни протеини могат да съдържат различни видовепорфирини (вижте точка 6). Хемът на хемоглобина съдържа протопорфирин IX, който има 4 метилови, 2 винилови радикала и 2 остатъка от пропионова киселина. Желязото в хема е в редуцирано състояние (Fe +2) и е свързано с две ковалентни и две координационни връзки с азотните атоми на пироловите пръстени. По време на окисляването на желязото хемът се преобразува
в хематин (Fe3+). Най-голямо количество хем се съдържа в червените кръвни клетки, пълни с хемоглобин, мускулните клетки, съдържащи миоглобин, и чернодробните клетки поради високото съдържание на цитохром Р 450.
b. биосинтеза на хема
Хемът се синтезира във всички тъкани, но най-често в костния мозък и черния дроб (Фиг. 13-2). В костния мозък хемът е необходим за синтеза на хемоглобин в ретикулоцитите, а в хепатоцитите - за образуването на цитохром Р 450.
Първата реакция на синтеза на хема - образуването на 5-аминолевулинова киселина от глицин и сукцинил-CoA (фиг. 13-3) се случва в митохондриалната матрица, където един от субстратите на тази реакция - сукцинил-CoA - се образува в TCA цикъл. Тази реакция се катализира от пиридоксал-зависимия ензим 5-аминолевулинатна синтаза.
От митохондриите 5-аминолевулиновата киселина навлиза в цитоплазмата. Междинните етапи на синтеза на хема се извършват в цитоплазмата: комбинацията от 2 молекули 5-аминолевулинова киселина в молекула порфобилиноген (фиг. 13-4), дезаминиране на порфобилиноген с образуването на хидроксиметилбилан, ензимно превръщане на хидроксиметилбилан в уропорфобилиноген III молекула, декарбоксилиране на последната с образуването на копропорфириноген III. Хидроксиметилбиланът може също да бъде неензимно преобразуван в уропорфириноген I, който се декарбоксилира в копропорфириноген I. От цитоплазмата копропорфириноген III отново навлиза в митохондриите, където протичат крайните реакции на синтеза на хема. В резултат на две поредни окислителни реакциикопропорфириноген III се превръща в протопорфириноген IX, а протопорфириноген IX се превръща в протопорфирин IX. Ензимът ферохелатаза, добавяйки двувалентно желязо към протопорфирин IX, го превръща в хем (фиг. 13-2). Източникът на желязо за синтеза на хема е протеинът феритин, съхраняващ желязо. син-
Ориз. 13-1. Структурата на порфин (A), протопорфирин IX (B) и хемоглобин хем (C).Порфинът е циклична структура, състояща се от четири пиролови пръстена, свързани с метенови мостове. Протопорфирин IX има четири метилови, два винилови и два остатъка на пропионова киселина. В хемоглобиновия хем Fe 2+ образува две ковалентни и две координационни връзки с азотните атоми на пироловите пръстени на протопорфирин IX.
Синтезираният хем, комбинирайки се с α- и β-полипептидните вериги на глобина, образува хемоглобин. Хемът регулира синтеза на глобин: когато скоростта на синтеза на хема намалява, синтезът на глобин в ретикулоцитите се инхибира.
Б. РЕГУЛИРАНЕ НА БИОСИНТЕЗАТА НА ХЕМ
Регулаторната реакция на синтеза на хема се катализира от пиридоксал-зависимия ензим 5-аминолевулинатна синтаза. Скоростта на реакцията се регулира алостерично и на ниво ензимна транслация.
Хемът е алостеричен инхибитор и корепресор на синтеза на 5-аминолевулинатна синтаза (фиг. 13-5).
В ретикулоцитите синтезата на този ензим на етапа на транслация се регулира от желязо. В мястото на иницииране на иРНК, кодираща ензима, има последователност от нуклеотиди, които образуват фибичка, която се нарича елемент, чувствителен към желязо (от английски. реагиращ на желязо елемент, IRE) (фиг. 13-6).
При високи концентрации на желязо в клетките, той образува комплекс с цистеинови остатъци на регулаторния желязо-свързващ протеин. Взаимодействието на желязото с регулаторния желязо-свързващ протеин причинява намаляване на афинитета на този протеин към IRE елемента на иРНК, кодираща 5-аминолевулинатна синтаза, и продължаване на транслацията (фиг. 13-6, А). При ниски концентрации на желязо, свързване на желязо

Ориз. 13-2. Синтез на хем.Числата на диаграмата показват ензимите: 1 - 5-аминолевулинат синтаза; 2 - 5-аминол-вулинат дехидратаза; 3 - порфобилиноген деаминаза; 4 - уропорфириноген III косинтаза; 5 - уропорфирин-гендекарбоксилаза; 6 - копропорфириноген III оксидаза; 7 - протопорфириноген оксидаза; 8 - ферохелатаза. Буквите означават заместители в пироловите пръстени: М - метил, В - винил, Р - остатъци от пропионова киселина, А - ацетил, PF - пиридоксал фосфат. Донор на желязо е протеинът феритин, който съхранява желязото в клетките.

Ориз. 13-3. Реакция на образуване на 5-аминолевулинова киселина.
протеинът се свързва с чувствителния към желязо елемент, разположен в 5" нетранслирания край на иРНК, и транслацията на 5-аминолевулинат синтазата се инхибира (фиг. 13-6, B).
5-аминолевулинат дехидратазата също се инхибира алостерично от хема, но тъй като активността на този ензим е почти 80 пъти по-висока от активността на 5-аминолевулинат синтазата, това е от малко физиологично значение.

Ориз. 13-4. Реакцията на образуване на порфобилиноген.

Ориз. 13-5. Регулиране на синтеза на хема и хемоглобина.Хемът, съгласно принципа на отрицателната обратна връзка, инхибира 5-аминолевулинат синтазата и 5-аминолевулинат дехидратазата и е индуктор на транслацията α- И β - хемоглобинови вериги.

Ориз. 13-6. Регулиране на синтеза на аминолевулин синтаза.А - при висока концентрацияжелязо в ретикулоцитите, то се свързва с желязо-свързващия протеин и намалява афинитета на този протеин към чувствителния към желязо елемент (IRE) на информационната РНК, кодираща 5-аминолевулинатна синтаза. Факторите за иницииране на транслацията на протеин се свързват с иРНК и инициират транслацията на 5-аминолевулинатна синтаза. B - с ниско съдържание на желязо в ретикулоцитите, желязо-свързващият протеин има висок афинитет към IRE и взаимодейства с него. Факторите за иницииране на транслацията на протеин не могат да се свържат с иРНК и транслацията спира.
Дефицит на пиридоксал фосфат и лекарства, които са негови структурни аналози, намаляват активността на 5-амино-левулинат синтазата.
Г. НАРУШЕНИЯ В БИОСИНТЕЗАТА НА ХЕМ. ПОРФИРИЯ
Наследствени и придобити нарушения на синтеза на хема, придружени от повишаване на съдържанието на порфириногени, както и техните продукти на окисление в тъканите и кръвта и появата им в урината, се наричат порфирии („порфирин“ в превод от гръцки означава лилаво). Урината на пациентите е червена.
Наследствените порфирии се причиняват от генетични дефекти в ензимите, участващи в
в синтеза на хема, с изключение на 5-аминолевулин синтазата. При тези заболявания се отбелязва намаляване на образуването на хем. Тъй като хемът е алостеричен инхибитор на 5-аминолевулинат синтазата, активността на този ензим се повишава и това води до натрупване на междинни продукти от синтеза на хема - 5-аминолевулинова киселина и порфириногени.
В зависимост от основното местоположение патологичен процесИма чернодробни и еритропоетични наследствени порфирии. Еритропоетичните порфирии се съпровождат от натрупване на порфирини в нормобластите и еритроцитите, а чернодробните порфирии - в хепатоцитите.
При тежки форми на порфирия се наблюдават нервно-психични разстройства, дисфункция на RES и увреждане на кожата. Порфириногените не са оцветени и не флуоресцират, но на светлина лесно се превръщат в порфирини. Последните проявяват интензивна червена флуоресценция в ултравиолетовите лъчи. В кожата на слънце, в резултат на взаимодействие с порфирините, кислородът преминава в синглетно състояние. Синглетният кислород ускорява липидната пероксидация на клетъчните мембрани и разрушаването на клетките, така че порфириите често са придружени от фоточувствителност и язви на откритите кожни участъци. Невропсихичните разстройства при порфириите са свързани с факта, че 5-аминолевулинатът и порфириногените са невротоксини.
Понякога при леки форми на наследствена порфирия заболяването може да бъде асимптоматично, но приемането на лекарства, които са индуктори на синтеза на 5-аминолевулинатна синтаза, може да доведе до обостряне на заболяването. Индуктори на синтеза на 5-аминолевулинатна синтаза са: известни лекарства, като сулфонамиди, барбитурати, диклофенак, волтарен, стероиди, гестагени. В някои случаи симптомите на заболяването не се появяват до пубертета, когато повишеното производство на β-стероиди предизвиква индукция на синтеза на 5-аминолевулинатна синтаза. Порфирия се наблюдава и при отравяне с оловни соли, тъй като оловото инхибира 5-аминолевулинат дехидратазата и ферохелатазата. Някои халоген-съдържащи хербициди и инсектициди са индуктори на синтеза на 5-аминолевулинатна синтаза, така че тяхното поглъщане е придружено от симптоми на порфирия.
II. МЕТАБОЛИЗЪМ НА ЖЕЛЯЗОТО
Тялото на възрастен човек съдържа 3-4 g желязо, от които само около 3,5 mg
намерени в кръвната плазма. Хемоглобинът съдържа приблизително 68% от желязото в цялото тяло, феритинът - 27%, миоглобинът - 4%, трансферинът - 0,1%, а всички желязосъдържащи ензими представляват само 0,6% от желязото, налично в тялото. Източниците на желязо в биосинтезата на желязосъдържащи протеини са желязото от храната и желязото, отделено при непрекъснатото разграждане на червените кръвни клетки в клетките на черния дроб и далака.
В неутрална или алкална среда желязото е в окислено състояние - Fe 3+, образувайки големи, лесно агрегиращи комплекси с OH -, други аниони и вода. При ниски стойности на pH желязото се редуцира и лесно се дисоциира. Процесът на редукция и окисление на желязото осигурява преразпределението му между макромолекулите в организма. Железните йони имат висок афинитет към много съединения и образуват хелатни комплекси с тях, променяйки свойствата и функциите на тези съединения, поради което транспортирането и отлагането на желязо в тялото се извършва от специални протеини. В клетките желязото се отлага от протеина феритин, а в кръвта се транспортира от протеина трансферин.
А. абсорбция на желязо в червата
В храната желязото е предимно в окислено състояние (Fe 3+) и е част от протеини или соли органични киселини. Освобождаването на желязо от соли на органични киселини се улеснява от киселата среда на стомашния сок. Най-голямо количество желязо се абсорбира в дванадесетопръстника. Аскорбинова киселина, съдържащ се в храната, възстановява желязото и подобрява усвояването му, тъй като само Fe 2+ навлиза в клетките на чревната лигавица. Дневното количество храна обикновено съдържа 15-20 mg желязо, като само около 10% от това количество се усвоява. Тялото на възрастен губи около 1 mg желязо на ден.
Количеството желязо, което се абсорбира в клетките на чревната лигавица, обикновено надвишава нуждите на организма. Доставката на желязо от ентероцитите в кръвта зависи от скоростта на синтеза на протеина апоферитин в тях. Апоферитинът "улавя" желязото в ентероцитите и се превръща във феритин, който остава в ентероцитите. По този начин потокът от
прехвърляне на желязо в кръвоносните капиляри от чревните клетки. Когато нуждата от желязо е ниска, скоростта на синтеза на апоферитин се увеличава (вижте по-долу „Регулиране на навлизането на желязо в клетките“). Постоянното ексфолиране на клетките на лигавицата в чревния лумен освобождава тялото от излишното желязо. При липса на желязо в организма апоферитинът почти не се синтезира в ентероцитите. Желязото, влизащо в кръвта от ентероцитите, транспортира кръвния плазмен протеин трансферин (фиг. 13-7).
Б. ТРАНСПОРТ НА ЖЕЛЯЗО В КРЪВНА ПЛАЗМА И НАВЛИЗАНЕТО МУ В КЛЕТКИТЕ
В кръвната плазма желязото се транспортира от протеина трансферин. Трансферинът е гликопротеин, който се синтезира в черния дроб и свързва само окисленото желязо (Fe 3+). Желязото, постъпващо в кръвта, се окислява от ензима фероксидаза, известен като съдържащия мед протеин на кръвната плазма церулоплазмин. Една молекула трансферин може да свърже един или два Fe 3+ йона, но едновременно с CO 3 2- аниона, за да образува трансферин-2 комплекс (Fe 3+ -CO 3 2-). Обикновено кръвният трансферин е наситен с желязо с приблизително 33%.
Трансферинът взаимодейства със специфични рецептори на клетъчната мембрана.
В резултат на това взаимодействие в цитозола на клетката се образува Ca 2+ -калмодулин-PKS комплекс, който фосфорилира транс-фериновия рецептор и предизвиква образуването на ендозома. ATP-зависимата протонна помпа, разположена в мембраната на ендозома, създава кисела среда вътре в ендозома. В киселата среда на ендозома желязото се освобождава от трансферина. След това комплексът рецептор-апотрансферин се връща на повърхността на плазмената мембрана на клетката. При неутрално рН на екстрацелуларната течност апотрансферинът променя своята конформация, отделя се от рецептора, навлиза в кръвната плазма и отново става способен да свързва железни йони и да се включва в нов цикъл на транспорта си в клетката. Желязото в клетката се използва за синтеза на желязосъдържащи протеини или се отлага в протеина феритин.
Феритинът е олигомерен протеин с молекулно тегло 500 kDa. Състои се от тежки (21 kDa) и леки (19 kDa) полипептидни вериги, включващи 24 протомера. Различният набор от протомери във феритиновия олигомер определя образуването на няколко изоформи на този протеин в различни тъкани. Феритинът е куха сфера, която може да съдържа до 4500 железни йони, но обикновено съдържа по-малко от 3000. Тежки вериги

Ориз. 13-7. Получаване на екзогенно желязо в тъканите.В чревната кухина желязото се освобождава от протеини и соли на органични киселини в храната. Аскорбиновата киселина, която намалява желязото, насърчава усвояването на желязото. В клетките на чревната лигавица излишното постъпващо желязо се свързва с протеина апоферитин, за да образува феритин, докато феритинът окислява Fe2+ до Fe3+. Навлизането на желязо от клетките на чревната лигавица в кръвта се придружава от окисление на желязото от ензима фероксидаза в кръвния серум. В кръвта Fe3+ се транспортира от серумния протеин трансферин. В тъканите Fe2+ се използва за синтеза на желязосъдържащи протеини или се отлага във феритин.
феритинът се окислява от Fe 2+ до Fe 3+ . Желязото под формата на хидроксид фосфат е разположено в центъра на сферата, чиято обвивка е образувана от протеиновата част на молекулата. То навлиза и се освобождава през канали, които проникват през протеиновата обвивка на апоферитина, но желязото може да се отложи и в протеиновата част на молекулата на феритина. Феритинът се намира в почти всички тъкани, но в най-големи количества в черния дроб, далака и костния мозък. Малка част от феритина се екскретира от тъканите в кръвната плазма. Тъй като навлизането на феритин в кръвта е пропорционално на съдържанието му в тъканите, концентрацията на феритин в кръвта е важен диагностичен показател за резервите на желязо в организма по време на желязодефицитна анемия. Метаболизмът на желязото в организма е показан на фиг. 13-8.
Б. РЕГУЛИРАНЕ НА ЖЕЛЯЗОТО В КЛЕТКИТЕ
Съдържанието на желязо в клетките се определя от съотношението на скоростите на неговия прием, използване и отлагане и се контролира от два молекулярни механизма. Скоростта на навлизане на желязото в нееритроидните клетки зависи от броя на трансфериновите рецепторни протеини в тяхната мембрана. Излишното желязо в клетките отлага феритин. Синтезът на рецепторите за апоферитин и трансферин се регулира на нивото на транслацията на тези протеини и зависи от съдържанието на желязо в клетката.
В нетранслирания 3" край на иРНК на трансфериновия рецептор и в нетранслирания 5" край на апоферитиновата иРНК има бримки на фиби - чувствителни към желязо елементи IRE (фиг. 13-9 и 13-10). Освен това, мРНК на трансферния рецептор

Ориз. 13-8. Метаболизъм на желязото в организма.

Ориз. 13-9. Регулиране на синтеза на апоферитин.А - когато съдържанието на желязо в клетката намалее, желязо-свързващият протеин има висок афинитет към IRE и взаимодейства с него. Това предотвратява свързването на факторите за иницииране на транслацията на протеин към иРНК, кодираща апоферитин, и синтезът на апоферитин спира; B - когато съдържанието на желязо в клетката се увеличи, то взаимодейства с желязо-свързващия протеин, в резултат на което афинитетът на този протеин към IRE намалява. Факторите за иницииране на транслацията на протеин се свързват с иРНК, кодираща апоферитин, и инициират транслацията на апоферитин.
Rin има 5 бримки, но апоферитиновата иРНК има само 1.
Тези области на иРНК могат да взаимодействат с регулаторния IRE-свързващ протеин. При ниски концентрации на желязо в клетката, IRE-свързващият протеин се свързва с IRE на апоферитин иРНК и предотвратява прикрепването на факторите за иницииране на транслацията на протеин (фиг. 13-9, A). В резултат скоростта на транслация на апоферитина и съдържанието му в клетката намаляват. В същото време, при ниски концентрации на желязо в клетката, IRE-свързващият протеин се свързва с чувствителния към желязо елемент на иРНК на трансфериновия рецептор и предотвратява разрушаването му от ензима РНКаза (фиг. 13-10, А). Това води до увеличаване на броя на рецепторите
трансферин и ускоряване навлизането на желязото в клетките.
Когато съдържанието на желязо в клетката се увеличи, в резултат на взаимодействието му с IRE-свързващия протеин, SH групите на активния център на този протеин се окисляват и афинитетът към чувствителните към желязо елементи на иРНК намалява. Това има две последици:
Първо, транслацията на апоферитин се ускорява (фиг. 13-9, B);
На второ място, IRE-свързващият протеин освобождава фибите на иРНК на трансфериновия рецептор и се унищожава от ензима РНКаза, в резултат на което скоростта на синтез на трансферинови рецептори намалява (фиг. 1310, B). Ускоряване на синтеза на апоферитин и инхибиране на трансрецепторния синтез

Ориз. 13-10. Регулиране на синтеза на трансферин рецептор.А - с ниско съдържание на желязо в клетката, чувствителният към желязо протеин има висок афинитет към IRE иРНК, кодираща протеина на рецептора на транс-ферин. Прикрепването на желязо-свързващия протеин към IRE иРНК предотвратява разрушаването му от РНКаза и синтезата на трансфериновия рецепторен протеин продължава; Б - Кога високо съдържаниежелязо в клетката, афинитетът на желязо-свързващия протеин към IRE намалява и иРНК става достъпна за действието на РНКаза, която я хидролизира. Разрушаването на иРНК води до намаляване на синтеза на протеина на трансфериновия рецептор.
ферин причинява намаляване на съдържанието
желязо в клетката. Като цяло тези механизми регулират съдържанието на желязо в клетките и използването му за синтеза на желязосъдържащи протеини.
Г. НАРУШЕНИЯ В МЕТАБОЛИЗМА НА ЖЕЛЯЗОТО
Желязодефицитна анемия може да възникне при повтарящи се кръвотечения, бременност, често раждане, язви и тумори
Стомашно-чревен тракт, след операции на стомашно-чревния тракт. При желязодефицитна анемия размерът на червените кръвни клетки и тяхната пигментация намаляват (малки хипохромни червени кръвни клетки). Съдържанието на хемоглобин в еритроцитите намалява, насищането на трансферин с желязо намалява и концентрацията на феритин в тъканите намалява. Причината за тези промени е липсата на желязо в организма, в резултат на което синтезът на хем и феритин в нееритроидните тъкани и хемоглобин в еритроидните клетки намалява.
Хемохроматоза.Когато количеството желязо в клетките превиши обема на феритиновото депо, желязото се отлага в протеиновата част на феритиновата молекула. В резултат на образуването на такива аморфни отлагания от излишното желязо феритинът се превръща в хемосидерин. Хемосидеринът е слабо разтворим във вода и съдържа до 37% желязо. Натрупването на гранули хемосидерин в черния дроб, панкреаса и далака води до увреждане на тези органи - хемохроматоза. Хемохроматозата може да бъде причинена от наследствено повишена абсорбция на желязо в червата, докато съдържанието на желязо в тялото на пациентите може да достигне 100 g. Това заболяване се наследява по автозомно-рецесивен начин и около 0,5% от кавказците са хомозиготни за генът на хемохроматозата. Натрупването на хемосидерин в панкреаса води до разрушаване на β-клетките на островите на Лангерханс и, като следствие, до захарен диабет. Отлагането на хемосидерин в хепатоцитите причинява цироза на черния дроб, а в миокардиоцитите - сърдечна недостатъчност. Пациентите с наследствена хемохроматоза се лекуват с редовно кръвопускане, седмично или веднъж месечно, в зависимост от тежестта на състоянието на пациента. Честите кръвопреливания могат да доведат до хемохроматоза; в тези случаи пациентите се лекуват с лекарства, които свързват желязото.
III. КАТАБОЛИЗЪМ НА ХЕМОГЛОБИН
Червените кръвни клетки имат кратко времеживот (приблизително 120 дни). При физиологични условия около 1-2x10 червени кръвни клетки на ден се разрушават в тялото на възрастен. Техният катаболизъм протича главно в ретикулоендотелните клетки на далака, лимфни възли, костен мозък и черен дроб. Със стареенето на еритроцитите съдържанието на сиалови киселини в състава на гликопротеините на плазмената мембрана намалява. Променените въглехидратни компоненти на гликопротеините на мембраните на еритроцитите се свързват с рецепторите на RES клетките и еритроцитите се „потапят“ в тях чрез ендоцитоза. Разграждането на червените кръвни клетки в тези клетки започва с разпадането на хемоглобина на хем и глобин и последваща хидролиза на протеиновата част на хемоглобина от лизозомни ензими.
А. ХЕМ КАТАБОЛИЗЪМ
Първата реакция на катаболизма на хема протича с участието на NADPH-зависимия ензимен комплекс хем оксигеназа.Ензимната система е локализирана в ER мембраната, в областта на електронните транспортни вериги на микрозомалното окисление. Ензимът катализира разцепването на връзката между два пиролови пръстена, съдържащи винилови остатъци, като по този начин разкрива структурата на пръстена (фиг. 13-11). По време на реакцията се образува линеен тетрапирол - биливердин(жълт пигмент) и въглероден оксид (CO), който се получава от въглерода на метенилната група. Хемът индуцира транскрипция на гена хемоксигеназа, който е абсолютно специфичен за хема.
Железните йони, освободени по време на разграждането на хема, могат да се използват за синтеза на нови молекули на хемоглобина или за синтеза на други протеини, съдържащи желязо. Биливердин се редуцира до билирубин от NADPH-зависим ензим. биливердин редуктаза.Билирубинът се образува не само от разграждането на хемоглобина, но и от катаболизма на други протеини, съдържащи хем, като цитохроми и миоглобин. Разграждането на 1 g хемоглобин произвежда 35 mg билирубин и приблизително 250-350 mg билирубин на ден при възрастен. По-нататъшният метаболизъм на билирубина се извършва в черния дроб.
Б. МЕТАБОЛИЗЪМ НА БИЛИРУБИН
Билирубинът, образуван в клетките на RES (далак и костен мозък), е слабо разтворим във вода и се транспортира през кръвта в комбинация с протеина на кръвната плазма албумин. Тази форма на билирубин се нарича неконюгиран билирубин. Всяка молекула албумин свързва 2 (или дори 3) молекули билирубин, една от които е свързана с протеина по-здраво (по-висок афинитет) от останалите. Когато pH на кръвта се измества към киселинната страна (повишена концентрация на кетонни тела, лактат), зарядът и конформацията на албумина се променят и афинитетът към билирубина намалява. Следователно билирубинът, който е слабо свързан с албумина, може да бъде изместен от свързващите центрове и да образува комплекси с колагена на междуклетъчния матрикс и мембранните липиди. Редица лекарствени съединения се конкурират с билирубина за

Ориз. 13-11. Разпадане на хема.М-(-СН3)-метилова група; В-(-СН=СН2)- винилова група; P-(-CH 2 -CH 2 -COOH) е остатъкът от пропионовата киселина. По време на реакцията една метилова група се превръща във въглероден оксид и по този начин отваря пръстенната структура. Полученият биливердин се превръща в билирубин под действието на биливердин редуктазата.
високоафинитетен, високоафинитетен център на албумин.
Поемане на билирубин от чернодробните паренхимни клетки
Комплексът албумин-билирубин, доставен чрез кръвния поток до черния дроб, на повърхността
Дебелината на плазмената мембрана на хепатоцита се дисоциира. Освободеният билирубин образува временен комплекс с липидите на плазмената мембрана. Улеснената дифузия на билирубин в хепатоцитите се осъществява от два вида протеини-носители: лигандин (пренася основното количество жлъчка-
рубин) и протеин Z. Активността на усвояването на билирубина от хепатоцита зависи от скоростта на неговия метаболизъм в клетката.
Лигандин и протеин Z също се намират в клетките на бъбреците и червата, следователно, в случай на недостатъчна чернодробна функция, те са в състояние да компенсират отслабването на процесите на детоксикация в този орган.
Конюгация на билирубин в гладката ER
В гладкия ER на хепатоцитите остатъците от глюкуронова киселина се добавят към билирубина - реакция на конюгация. Билирубинът има 2 карбоксилни групи, така че може да се комбинира с 2 молекули глюкуронова киселина, образувайки силно водоразтворим конюгат - билирубин ди-глюкуронид (конюгиран или директен билирубин) (фиг. 13-12).
Донорът на глюкуроновата киселина е UDP-глюкуронат. Специфични ензими, UDP-глюкуронилтрансферази (уридин дифосфоглюкуронилтрансферази) катализират образуването на билирубинови моно- и диглюкурониди (фиг. 13-13). Някои лекарства, например фенобарбитал, са индуктори на синтеза на UDP-глюкуронилтрансферази (вижте точка 12).
Секреция на билирубин в жлъчката
Секрецията на конюгиран билирубин в жлъчката става чрез механизма на активен транспорт, т.е. срещу концентрационен градиент. Активният транспорт вероятно е етапът, ограничаващ скоростта на целия процес на метаболизма на билирубина в черния дроб. Обикновено диглюкуронид

Ориз. 13-12. Структура на билирубин диглюкуронид (конюгиран, "директен" билирубин).Глюкуроновата киселина се свързва с естерна връзка с два остатъка на пропионова киселина, за да образува ацил глюкуронид.
билирубинът е основната форма на екскреция на билирубина в жлъчката, но не може да се изключи наличието на малко количество моно-глюкуронид. Транспортирането на конюгиран билирубин от черния дроб до жлъчката се активира от същите лекарства, които могат да индуцират конюгацията на билирубин. По този начин можем да кажем, че скоростта на конюгация на билирубина и активния транспорт на билирубинов глюкуронид от хепатоцитите до жлъчката са тясно взаимосвързани (фиг. 13-14).
Б. КАТАБОЛИЗЪМ НА БИЛИРУБИН ДИГЛЮКУРОНИД
В червата входящите билирубинови глюкурониди се хидролизират от специфични бактериални ензими β-глюкуронидази, които хидролизират връзката между билирубина и остатъка от глюкуронова киселина. Освободен

Ориз. 13-13. Образуване на билирубин диглюкуронид.

Ориз. 13-14. Цикъл на билирубин-уробилиноген в черния дроб. 1 - катаболизъм на Hb в ретикулоендотелните клетки на костния мозък, далака, лимфните възли; 2 - образуване на транспортната форма на комплекса билирубин-албумин; 3 - навлизане на билирубин в черния дроб; 4 - образуване на билирубинови глюкурониди;
5 - секреция на билирубин като част от жлъчката в червата;
6 - катаболизъм на билирубина под влияние чревни бактерии; 7 - отстраняване на уробилиногени с изпражнения; 8 - абсорбция на уробилиногени в кръвта; 9 - усвояване на уробилиногени от черния дроб; 10 - навлизане на някои уробилиногени в кръвта и екскреция от бъбреците с урината; 11 - малка част от уробилиногените се секретира в жлъчката.
Билирубинът, произведен по време на тази реакция, се редуцира под въздействието на чревната микрофлора, за да образува група от безцветни тетрапиролни съединения - уробилиногени(фиг. 13-15).
В илеума и дебелото черво малка част от уробилиногените се реабсорбират и навлизат в черния дроб с кръвта на порталната вена. Основната част от уробилиногена от черния дроб като част от жлъчката се екскретира в червата и се екскретира от
отделяйки се от тялото, част от уробилиногена от черния дроб навлиза в кръвта и се елиминира с урината под формата на уробилин (фиг. 13-14). Обикновено повечето безцветни уробилиногени, образувани в дебелото черво, се окисляват в ректума до пигмент под въздействието на чревната микрофлора кафяво уробилини се елиминира с изпражненията. Цветът на изпражненията се дължи на наличието на уробилин.

Ориз. 13-15.Структура на някои жлъчни пигменти. Мезобилиногенът е междинен продукт от катаболизма на билирубина в червата.
iv. диагностична стойност
определяне на концентрацията на билирубин в човешките биологични течности
Понастоящем за определяне на съдържанието на билирубин в кръвния серум (плазма) се използва методът, предложен през 1916 г. от Ван дер Берг за определяне на билирубин в кръвния серум, базиран на диазореакцията.
Нормална концентрация общ билирубинв плазмата е 0,3-1 mg/dl (1,7-17 µmol/l), 75% от общото количество билирубин е в неконюгирана форма (индиректен билирубин). В клиниката конюгираният билирубин се нарича директен билирубин, тъй като е водоразтворим и може бързо да реагира с диазореагента, образувайки розово съединение - това е директната реакция на Ван дер Берг. Неконюгираният билирубин е хидрофобен, поради което се намира в кръвната плазма в комплекс с албумин и не реагира с диазореагента, докато не се добави органичен разтворител, като етанол, който утаява албумина. Неконюгираният билирубин, който реагира с азобагрилото само след утаяване на протеина, се нарича индиректен билирубин.
При пациенти с хепатоцелуларна патология, придружена от дългосрочно увеличение
концентрации на конюгиран билирубин, в кръвта се открива трета форма на плазмен билирубин, при която билирубинът е ковалентно свързан с албумина и следователно не може да бъде отделен по обичайния начин. В някои случаи до 90% от общото съдържание на билирубин в кръвта може да бъде в тази форма.
А. ЖЪЛТЕНИЦА
Причините за хипербилирубинемия могат да бъдат повишено производство на билирубин, което надвишава способността на черния дроб да го отделя, или увреждане на черния дроб, което води до нарушаване на секрецията на билирубин в жлъчката в нормални количества. Хипербилирубинемия също се отбелязва, когато жлъчните пътища на черния дроб са блокирани.
Във всички случаи съдържанието на общ билирубин в кръвта се повишава. Когато се достигне определена концентрация, той дифундира в тъканите, оцветявайки ги в жълто. Пожълтяването на тъканите поради отлагането на билирубин в тях се нарича жълтеница. Клинично жълтеницата може да не се появи, докато концентрацията на билирубин в кръвната плазма не превиши горната граница на нормата повече от 2,5 пъти, т.е. няма да се повиши над 50 µmol/l.
1. Хемолитична (супрахепатална) жълтеница
Известно е, че способността на черния дроб да образува глюкурониди и да ги отделя в жлъчката е 3-4 пъти по-висока от образуването им при физиологични условия. Хемолитичната (супрахепатална) жълтеница е резултат от интензивна хемолиза на червените кръвни клетки. Причинява се от прекомерно образуване на билирубин, превишаване
способността на черния дроб да го отделя. Хемолитичната жълтеница се развива при изчерпване на резервния капацитет на черния дроб. Основната причина за супрахепаталната жълтеница е наследствена или придобита хемолитична анемия. При хемолитична анемия, причинена от сепсис, лъчева болест, дефицит на глюкозо-6-фосфат дехидрогеназа на еритроцитите, таласемия, трансфузия на несъвместими кръвни групи, отравяне със сулфонамиди, количеството хемоглобин, освободен от еритроцитите на ден, може да достигне до 45 g
(при норма 6,25 g), което значително повишава образуването на билирубин. Хипербилирубинемията при пациенти с хемолитична жълтеница се причинява от значително повишаване (103-171 µmol/l) в кръвната концентрация на албумин-свързан неконюгиран билирубин (индиректен билирубин). Образуването в черния дроб и навлизането в червата на големи количества билиру-бинглюкурониди (директен билирубин) води до повишено образуване и екскреция на уробилиногени в изпражненията и урината и тяхното по-интензивно оцветяване (фиг. 13-16).
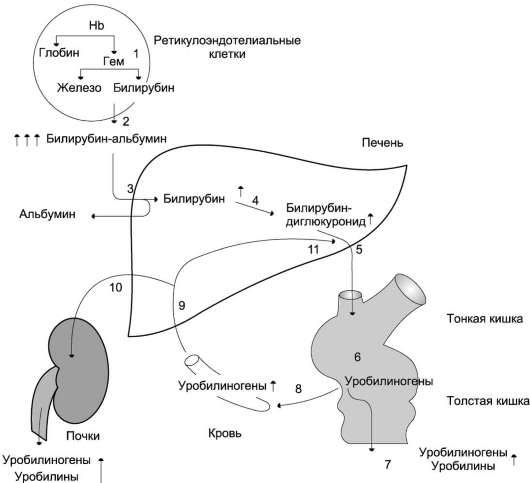
Ориз. 13-16. Цикъл на билирубин-уробилиноген при хемолитична жълтеница. 1 - Hb катаболизмът идва с повишена скорост; 2 - концентрацията в кръвта се увеличава приблизително 10 пъти директен билирубин; 3 - албуминът се освобождава от комплекса билирубин-албумин; 4 - активността на реакцията на глюкурониране се увеличава, но е по-ниска от скоростта на образуване на билирубин; 5 - повишена секреция на билирубин в жлъчката; 6, 7,10 - повишеното съдържание на уробилиногени в изпражненията и урината им придава по-интензивен цвят; Уробилиногенът се абсорбира от червата в кръвта (8) и навлиза в черния дроб през порталната вена (9).
Един от основните признаци хемолитична жълтеница- повишени нива на неконюгиран (индиректен) билирубин в кръвта. Това улеснява разграничаването й от механична (субхепатална) и хепатоцелуларна (черен дроб) жълтеница.
Неконюгираният билирубин е токсичен. Хидрофобен, липофилен неконюгиран билирубин, който лесно се разтваря в мембранните липиди и по този начин прониква в митохондриите, разединява дишането и окислителното фосфорилиране в тях, нарушава протеиновия синтез, потока на калиеви йони през мембраната на клетката и органелите. Това се отразява негативно на състоянието на централната нервна система, причинявайки редица характерни неврологични симптоми при пациентите.
Жълтеница на новородени
Особен вид хемолитична жълтеница при новородени е "физиологична жълтеница", която се наблюдава в първите дни от живота на детето. Причината за повишаване на концентрацията индиректен билирубинв кръвта има ускорена хемолиза и недостатъчност на функцията на протеини и чернодробни ензими, отговорни за абсорбцията, конюгацията и секрецията на директния билирубин. При новородени не само активността на UDP-глюкуронилтрансферазата е намалена, но също така, очевидно, синтезът на втория субстрат на реакцията на конюгиране на UDP-глюкуронат не е достатъчно активен.
Известно е, че UDP-глюкуронилтрансферазата е индуцируем ензим (вижте точка 12). На новородени с физиологична жълтеница се прилага лекарството фенобарбитал, чийто индуциращ ефект е описан в точка 12.
Едно от неприятните усложнения " физиологична жълтеница" - билирубинова енцефалопатия. Когато концентрацията на неконюгиран билирубин надвиши 340 µmol/L, той преминава през кръвно-мозъчната бариера и причинява увреждане на мозъка.
2. Хепатоцелуларна (чернодробна) жълтеница
Хепатоцелуларната (чернодробна) жълтеница се причинява от увреждане на хепатоцитите и жлъчните капиляри, например при остри вирусни инфекции, хроничен и токсичен хепатит.
Причината за повишаване на концентрацията на билирубин в кръвта е увреждане и некроза на някои чернодробни клетки. Има задържане на билирубин в черния дроб, което се улеснява от рязкото отслабване на метаболитните процеси в засегнатите хепатоцити, които губят способността си да изпълняват нормално различни биохимични и физиологични функции, по-специално да прехвърлят конюгиран (директен) билирубин от клетките в жлъчката срещу концентрационен градиент. Характерното за хепатоцелуларната жълтеница е, че вместо нормално преобладаващите билирубинови диглюкурониди се образуват моноглюкурониди главно в засегнатата чернодробна клетка
(фиг. 13-17).
В резултат на разрушаването на чернодробния паренхим, полученият директен билирубин частично навлиза в голям кръгкръвообращението, което води до жълтеница. Отделянето на жлъчка също е нарушено. В червата навлиза по-малко билирубин от нормалното.
При чернодробно-клетъчна жълтеница се повишава концентрацията в кръвта както на общия билирубин, така и на двете му фракции - неконюгиран (индиректен) и конюгиран (директен).
Тъй като по-малко билирубин глюкуронид навлиза в червата, количеството на произведения уробилиноген също намалява. Следователно изпражненията са хипохолични, т.е. по-малко оцветен. Урината, напротив, има по-интензивен цвят поради наличието не само на уробилини, но и на конюгиран билирубин, който е силно разтворим във вода и се екскретира в урината.
3. Механична или обструктивна (субхепатална) жълтеница
Механична или обструктивна (чернодробна) жълтеница се развива, когато жлъчната секреция е нарушена в дванадесетопръстника. Това може да бъде причинено от запушване на жлъчните пътища, например поради холелитиаза, тумор на панкреаса, жлъчен мехур, черен дроб, дванадесетопръстник, хронично възпалениепанкреас или следоперативно стеснение на общия жлъчен канал (фиг. 13-18).
В случай на пълно запушване на общия жлъчен канал, конюгиран билирубин в състава

Ориз. 13-17. Нарушение на билирубин-уробилиногеновия цикъл при хепатоцелуларна жълтеница.В черния дроб скоростта на реакцията на глюкурониране на билирубин е намалена (4), поради което концентрацията на индиректен билирубин в кръвта се увеличава; Поради нарушение на чернодробния паренхим, част от билирубиновия глюкуронид, образуван в черния дроб, навлиза в кръвта (12) и след това се отстранява от тялото с урината (10). В урината на пациентите присъстват уробилини и билирубинови глюкурониди. Останалите числа съответстват на етапите на метаболизма на билирубина на фиг. 13-16.
жлъчката не навлиза в червата, въпреки че хепатоцитите продължават да я произвеждат. Тъй като билирубинът не навлиза в червата, в урината и изпражненията няма продукти от неговия катаболизъм, уробилиногени. Изпражненията са обезцветени. Тъй като нормалните пътища на отделяне на билирубина са блокирани, той изтича в кръвта, така че концентрацията на конюгиран билирубин в кръвта на пациентите се повишава. Разтворимият билирубин се екскретира с урината, придавайки й наситен оранжево-кафяв цвят.
Б. ДИФЕРЕНЦИАЛНА ДИАГНОЗА НА ЖЪЛТЕНАТА
При диагностицирането на жълтеница трябва да се има предвид, че на практика жълтеница от който и да е вид рядко се отбелязва в „чиста“ форма. По-често има комбинация от един или друг тип. По този начин, при тежка хемолитична жълтеница, придружена от повишаване на концентрацията на индиректен билирубин, различни органи неизбежно страдат, включително черния дроб, който може да въведе елементи

Ориз. 13-18. Нарушение на билирубин-уробилиногеновия цикъл при обструктивна жълтеница.Поради обструкция на жлъчния мехур билирубин глюкуронидът не се секретира в жлъчката (5); липсата на билирубин в червата води до обезцветяване на изпражненията (6); разтворимият билирубин глюкуронид се екскретира от бъбреците в урината (10). В урината липсват уробилини; Образуваният в черния дроб билирубинов глюкуронид навлиза в кръвта (12), в резултат на което съдържанието на директен билирубин се увеличава. Останалите числа съответстват на етапите на метаболизма на билирубина на фиг. 13-16.
паренхимна жълтеница, т.е. повишен директен билирубин в кръвта и урината. От своя страна, паренхимната жълтеница, като правило, включва елементи на механична жълтеница. При чернодробна (обструктивна) жълтеница, например при рак на главата на панкреаса, повишената хемолиза е неизбежна в резултат на ракова интоксикация и, като следствие, повишаване на директния и индиректния билирубин в кръвта.
Така че хипербилирубинемията може да бъде следствие от излишък както на свързани, така и на свободни
билирубин. Измерването на техните концентрации поотделно е необходимо при диагностициране на жълтеница. Ако концентрацията на билирубин в плазмата<100 мкмоль/л и другие тесты функции печени дают нормальные результаты, возможно предположить, что повышение обусловлено за счёт непрямого билирубина. Чтобы подтвердить это, можно сделать анализ мочи, поскольку при повышении концентрации непрямого билирубина в плазме прямой билирубин в моче отсутствует.
При диференциалната диагноза на жълтеницата е необходимо да се вземе предвид съдържанието на уробилиногени в урината. Обикновено около 4 mg уробилиногени се екскретират от тялото с урината на ден. Ако в урината се отделя повишено количество уробилиногени, това е доказателство за чернодробна недостатъчност, например с чернодробна или хемолитична жълтеница. Наличието в урината не само на уробилиногени, но и на директен билирубин показва увреждане на черния дроб и нарушено изтичане на жлъчка в червата.
Б. НАСЛЕДСТВЕНИ НАРУШЕНИЯ В МЕТАБОЛИЗМА НА БИЛИРУБИН
Има няколко известни заболявания, при които жълтеницата се причинява от наследствени нарушения на метаболизма на билирубина.
Приблизително 5% от населението е диагностицирано с наследствена жълтеница, причинена от генетични нарушения в структурата на протеини и ензими, отговорни за транспорта (усвояването) на индиректния билирубин в черния дроб и конюгирането му с глюкуронова киселина. Тази патология се наследява по автозомно-доминантен начин. Концентрацията на индиректен билирубин в кръвта на пациентите е повишена.
Има 2 вида наследствена жълтеница, причинена от нарушение на реакцията на глюкурониране в черния дроб - образуването на директен билирубин.
Първият тип се характеризира с пълна липса на UDP-глюкуронилтрансфераза. Аз ще-
Унаследява се по автозомно-рецесивен начин. Приложението на фенобарбитал, индуктор на UDP-глюкуронилтрансфераза, не води до намаляване на нивата на билирубина. Децата умират в ранна възраст поради развитието на билирубинова енцефалопатия.
Вторият тип се характеризира с намаляване на активността (недостатъчност) на UDP-глюкуронил трансферазата; хипербилирубинемията възниква поради индиректен билирубин. Жълтеницата се повлиява добре от лечение с фенобарбитал.
Нарушаването на активния транспорт на билирубинови глюкурониди, образувани в чернодробните клетки в жлъчката, е характерно за жълтеницата, наследена по автозомно-доминантен начин. Проявява се чрез хипербилирубинемия, дължаща се на директен билирубин и билирубинурия (директният билирубин се определя в урината).
Фамилната хипербилирубинемия при новородени се свързва с наличието на конкурентни инхибитори на конюгацията на билирубина (естрогени, свободни мастни киселини) в кърмата. По време на кърмене в кръвния серум на бебето се откриват инхибитори на конюгацията на билирубина. Такава хипербилирубинемия се нарича преходна. Хипербилирубинемията изчезва, когато детето премине на изкуствено хранене. Нелекуваната хипербилирубинемия води до развитие на билирубинова енцефалопатия и ранна смърт.