
Osteodystrophy ทางพันธุกรรมของออลไบรท์ Pseudohypoparathyroidism (โรคกระดูกพรุนทางพันธุกรรมของ Albright): ความยากลำบากในการค้นหาการวินิจฉัยแยกโรค การสังเกตทางคลินิก อาการ Pseudohypoparathyroidism
Pseudohypoparathyroidism
Pseudohypoparathyroidism คืออะไร -
Pseudohypoparathyroidism(กรีก pseudē เป็นเท็จ + ภาวะต่อมพาราไทรอยด์ต่ำ; คำพ้องความหมาย: โรคกระดูกเสื่อมทางพันธุกรรมของอัลไบรท์, โรคของอัลไบรท์) เป็นโรคทางพันธุกรรมที่หายาก ระบบโครงกระดูก, จำลองภาวะ hypoparathyroidism และโดดเด่นด้วยการเผาผลาญแคลเซียมและฟอสฟอรัสบกพร่อง; มักมาพร้อมกับพัฒนาการทางจิตใจและร่างกายที่ล่าช้า
อะไรกระตุ้น / สาเหตุของ Pseudohypoparathyroidism:
สาเหตุของ pseudohypoparathyroidism เป็นข้อบกพร่องที่มีมา แต่กำเนิด - ความไม่รู้สึกของเนื้อเยื่อส่วนปลายต่อการกระทำของ PTH
กลไกการเกิดโรค (จะเกิดอะไรขึ้น?) ระหว่าง Pseudohypoparathyroidism:
เป็นที่เชื่อกันว่า pseudohypoparathyroidism ขึ้นอยู่กับความต้านทานทางพันธุกรรมของไตและโครงกระดูกต่อการทำงานของฮอร์โมนพาราไธรอยด์อันเป็นผลมาจากข้อบกพร่องในตัวรับไซโตรีเซพเตอร์เฉพาะ - ฮอร์โมนพาราไธรอยด์ - อะดีนิเลตไซโคลสคอมเพล็กซ์ซึ่งขัดขวางการก่อตัวในไตของไซคลิก 3 ", 5"-AMP ซึ่งเป็นสื่อกลางในเซลล์ของการกระทำของฮอร์โมนพาราไธรอยด์ต่อกระบวนการเผาผลาญ . Pseudohypoparathyroidism เป็นโรคที่มีความหลากหลายทางพันธุกรรม ในผู้ป่วยบางราย ตัวรับไซโตรีเซพเตอร์เองที่จับกับฮอร์โมนพาราไธรอยด์มีข้อบกพร่อง (ประเภท Ia pseudohypoparathyroidism) ส่วนตัวอื่นๆ มีข้อบกพร่องในโปรตีนที่จับกับนิวคลีโอไทด์ ซึ่งแปลเป็นภาษาท้องถิ่นในไขมันสองชั้นของเยื่อหุ้มเซลล์และทำหน้าที่จับกับตัวรับกับอะดีนิเลตไซเคลส (ประเภท Ib pseudohypoparathyroidism) ผู้ป่วยบางรายมีประสบการณ์ การขาดเอนไซม์ adenylate cyclase นั่นเอง (pseudohypoparathyroidism type II) การขาดแคมป์ที่เกิดจากข้อบกพร่องเหล่านี้นำไปสู่การหยุดชะงักของการสังเคราะห์โปรตีนเฉพาะที่กำหนดผลกระทบทางชีวภาพของฮอร์โมนพาราไธรอยด์ ดังนั้นความไวของอวัยวะเป้าหมาย โดยเฉพาะไต ต่อฮอร์โมนพาราไธรอยด์จึงหายไป เป็นผลให้การขับถ่ายของฟอสฟอรัสในปัสสาวะลดลง, ภาวะฟอสเฟตในเลือดสูงเกิดขึ้นและภาวะแคลเซียมในเลือดต่ำจะพัฒนาเป็นลำดับที่สอง เนื่องจากใน pseudohypoparathyroidism ต่อมพาราไธรอยด์ยังคงเดิมอยู่ต่อมรองอาจพัฒนาขึ้นเพื่อตอบสนองต่อภาวะน้ำตาลในเลือดต่ำซึ่งจะช่วยกระตุ้นการผลิตฮอร์โมนพาราไธรอยด์ ภาวะต่อมพาราไทรอยด์ทำงานเกิน. การก่อตัวของฮอร์โมนพาราไธรอยด์ที่เพิ่มขึ้นไม่ทำให้เกิดการขับถ่ายของฟอสฟอรัสและแคมป์ในปัสสาวะเพิ่มขึ้นเนื่องจากการต้านทานทางพันธุกรรมของท่อไตต่อฮอร์โมนพาราไธรอยด์ แต่จะมาพร้อมกับการเปลี่ยนแปลงใน เนื้อเยื่อกระดูกลักษณะของภาวะพาราไธรอยด์เกินซึ่งบ่งชี้ถึงการรักษาความไวปกติของเซลล์สร้างกระดูกต่อฮอร์โมนพาราไธรอยด์ ในกิจกรรม pseudohypoparathyroidism อัลคาไลน์ฟอสฟาเตสในซีรั่มในเลือดเพิ่มขึ้นหรืออยู่ในขอบเขตปกติ (0.5-1.3 ไมโครโมลฟอสฟอรัสอนินทรีย์ต่อ 1 มลเซรั่มเลือดสำหรับ 1 ชม.การฟักตัวที่ 37°; คำจำกัดความตาม Bodansky) pseudohypoparathyroidism ทุกรูปแบบเป็นโรคทางพันธุกรรม โดยธรรมชาติของการถ่ายทอดทางพันธุกรรมจะมีลักษณะเด่นแบบออโตโซม ภาวะเจริญพันธุ์ต่ำของผู้ชายที่เป็นโรค pseudohypoparathyroidism อธิบายถึงความหายากของการถ่ายทอดจากพ่อสู่ลูก ผู้หญิงป่วยบ่อยกว่าผู้ชาย 2 เท่า
โดยปกติแล้วเมื่อมี pseudohypoparathyroidism จะพบว่ามีการชดเชยภาวะ hyperplasia ต่อมพาราไธรอยด์(การมีเนื้องอกในนั้นไม่ใช่เรื่องปกติ) การเปลี่ยนแปลงตามแบบฉบับของภาวะพาราไธรอยด์ในเลือดสูงจะถูกบันทึกไว้ในเนื้อเยื่อกระดูก - โรคกระดูกพรุนแบบกระจาย, การปรากฏตัวของซีสต์ (ที่เรียกว่าเนื้องอกสีน้ำตาล, เนื้องอกเซลล์ขนาดยักษ์) แคลเซียมที่ปล่อยออกมาจากกระดูกจะสะสมอยู่ในรูปของการกลายเป็นปูนใน เนื้อเยื่อใต้ผิวหนังรวมทั้งในไต กล้ามเนื้อ กล้ามเนื้อหัวใจ ผนัง หลอดเลือดแดงใหญ่, เยื่อบุตาและตามขอบกระจกตา
อาการของ Pseudohypoparathyroidism:
อาการทางคลินิกของ pseudohypoparathyroidism มีความคล้ายคลึงกับอาการที่ไม่ทราบสาเหตุ ภาวะต่อมพาราไทรอยด์ต่ำ. มีการโจมตีของยาชูกำลังชักที่เกิดขึ้นเองหรืออยู่ภายใต้อิทธิพลของการระคายเคือง การกลายเป็นปูนในเนื้อเยื่อใต้ผิวหนังมีแนวโน้มที่จะเป็นแผล ขบวนการสร้างกระดูกใต้ผิวหนังมักรุนแรงถึงขนาดที่เลียนแบบการสร้างกระดูกอักเสบ (myositis ossificans) ลักษณะเฉพาะ ได้แก่ ปัญญาอ่อน การเจริญเติบโตแคระ หน้าพระจันทร์ โรคอ้วน และ brachydactyly โดยเฉพาะอย่างยิ่งการลดขนาด metacarpals ที่ 1, 4 และ 5 และ metatarsals อาจสังเกตเห็น exostoses หลายครั้ง, dyschondroplasia, อาการต่างๆ ภาวะต่อมพาราไธรอยด์ในเลือดสูงทุติยภูมิในรูปแบบของการสลายของกระดูกนิ้ว subperiosteal; การเปลี่ยนแปลงใน epiphyses ของกระดูกจะเหมือนกับใน fibrous Osteodysplasia มักสังเกตเห็นการอาเจียนเช่นเดียวกับปัสสาวะเนื่องจากการก่อตัวของนิ่วออกซาเลตในทางเดินปัสสาวะ, ตรวจพบต้อกระจกแม่และเด็ก, และ hypoplasia ของเคลือบฟัน
ในคนไข้ที่เป็น pseudohypoparathyroidism พร้อมกับความไวของอวัยวะเป้าหมายต่อฮอร์โมนพาราไธรอยด์ลดลง อาจสังเกตความต้านทานต่อฮอร์โมนอื่น ๆ ที่ขึ้นอยู่กับระบบ adenylate cyclase เช่นอวัยวะสืบพันธุ์ต่อฮอร์โมน gonadotropic ต่อมไทรอยด์ต่อฮอร์โมนกระตุ้นต่อมไทรอยด์ , อวัยวะเป้าหมายไปที่ฮอร์โมนกลูคากอนและแอนติไดยูเรติก มีความถี่เพิ่มขึ้น โรคแพ้ภูมิตัวเองและ โรคเบาหวานสังเกตภาวะพร่องไทรอยด์และภาวะต่อมไทรอยด์ทำงานเกิน
นอกจากนี้ยังมี pseudopseudohypoparathyroidism ซึ่งเป็นลักษณะที่ไม่มีภาวะน้ำตาลในเลือดต่ำ, ภาวะฟอสเฟตในเลือดสูง, อาการชักและโรคกระดูกพรุน
การวินิจฉัยโรค Pseudohypoparathyroidism:
การวินิจฉัยในกรณีทั่วไปของโรคเกิดขึ้นในเด็กอายุ 5-10 ปีบนพื้นฐานของภาพทางคลินิกที่มีลักษณะเฉพาะ, ความผิดปกติหลายประการในการพัฒนาของโครงกระดูกกระดูก, การปรากฏตัวของภาวะน้ำตาลในเลือดต่ำ, ภาวะฟอสเฟตในเลือดสูง, กิจกรรมปกติหรือเพิ่มขึ้นของอัลคาไลน์ฟอสฟาเตส ในซีรั่มในเลือดลดการขับแคลเซียมและฟอสฟอรัสในปัสสาวะ เนื้อหาสูงฮอร์โมนพาราไธรอยด์ในเลือด การปรากฏตัวของความต้านทานท่อไตต่อฮอร์โมนพาราไธรอยด์ได้รับการยืนยันโดยการทดสอบโดยพิจารณาจากปริมาณฟอสเฟตและแคมป์ที่ถูกขับออกมาในปัสสาวะ การไม่มีเนื้อหาฟอสเฟตและแคมป์เพิ่มขึ้นอย่างมีนัยสำคัญในปัสสาวะหลังจากให้ฮอร์โมนพาราไธรอยด์แก่ผู้ป่วยแล้วบ่งชี้ว่าไตมีความต้านทานต่อการทำงานของฮอร์โมนพาราไธรอยด์ ในคนไข้ที่ไม่ทราบสาเหตุและหลังผ่าตัด ในทางกลับกัน หลังจากได้รับฮอร์โมนพาราไธรอยด์ 200 หน่วยทางหลอดเลือดดำในปัสสาวะ ปริมาณฟอสเฟตและแคมป์ภายใน 4 ชม.เพิ่มขึ้น 2-10 เท่าเมื่อเทียบกับ พื้นฐาน. การขับถ่ายของ hydroxyproline ในปัสสาวะในผู้ป่วยที่ไม่ได้รับการรักษาด้วย pseudohypoparathyroidism เป็นเรื่องปกติหรือเพิ่มขึ้นเล็กน้อยและใน hypoparathyroidism ก็ลดลง การวินิจฉัยด้วยรังสีเอกซ์ของ pseudohypoparathyroidism ขึ้นอยู่กับการระบุการเปลี่ยนแปลงเฉพาะในกระดูกและเนื้อเยื่ออ่อน
Pseudohypoparathyroidism ร่วมกับ hypogonadism ในเพศหญิงจะต้องมีความแตกต่างกัน Shereshevsky - กลุ่มอาการเทิร์นเนอร์ซึ่ง pseudopseudohypoparathyroidism มีความคล้ายคลึงกันทางฟีโนไทป์ ในกลุ่มอาการ Shereshevsky-Turner ไม่พบโครมาตินทางเพศ แทนที่รังไข่จะมีเส้นใยเนื้อเยื่อเกี่ยวพันซึ่งไม่สามารถตรวจพบได้ในระหว่างการตรวจทางทวารหนักและอัลตราซาวนด์
การรักษา Pseudohypoparathyroidism:
การรักษาภาวะแคลเซียมในเลือดต่ำประกอบด้วยการสั่งจ่ายแคลเซียมเสริมในปริมาณที่เพียงพอเพื่อรักษาความเข้มข้นของแคลเซียมในเลือดให้เป็นปกติ ความสำคัญอย่างยิ่งมีการบำบัดด้วยวิตามินดี ขนาดยาเริ่มแรกคำนวณจาก 2000 IU/ กิโลกรัมน้ำหนักตัวต่อวัน แต่ไม่เกิน 100,000 IU ต่อวัน เพื่อหลีกเลี่ยงการเตรียมวิตามินดีเกินขนาดจำเป็นต้องตรวจสอบความเข้มข้นของแคลเซียมในเลือดทุก 3-7 วันในช่วงสองสัปดาห์แรกของการรักษาและทุกเดือนในช่วง 2-3 เดือนข้างหน้า เมื่อความเข้มข้นของแคลเซียมในเลือดถึงระดับคงที่แล้ว ก็เพียงพอที่จะตรวจดูทุกๆ 2-3 เดือน คุณสามารถใช้แคลซิทริน, ไดไฮโดรทาคิสเตอรอล, ออกซีเดวิตและยาอื่น ๆ ได้ แบบฟอร์มที่ใช้งานอยู่วิตามินดี อาหารที่ถูกจำกัดด้วยฟอสฟอรัสช่วยให้ความเข้มข้นของแคลเซียมในเลือดเป็นปกติและกำจัดอาการของภาวะพาราไทรอยด์รอยด์ทุติยภูมิทุติยภูมิ ในกรณีที่ต่อมอื่นไม่เพียงพอ การหลั่งภายในดำเนินการบำบัดทดแทนด้วยฮอร์โมนที่เหมาะสม การรักษาด้วยฮอร์โมนพาราไธรอยด์ไม่ได้ผล เพื่อหยุดการโจมตีแบบชักจะมีการให้สารละลายแคลเซียมคลอไรด์หรือแคลเซียมกลูโคเนต 10% ทางหลอดเลือดดำ ทางปาก - สารละลายแคลเซียมคลอไรด์ 5-10%, 1 ช้อนโต๊ะ 3-4 ครั้งต่อวัน: แคลเซียมกลูโคเนต, แคลเซียมแลคเตต - มากถึง 10 ชในหนึ่งวัน.
การพยากรณ์โรคด้วยการบำบัดอย่างมีเหตุผลเป็นสิ่งที่ดี เมื่อพิจารณาถึงลักษณะทางพันธุกรรมของภาวะไทรอยด์ฮอร์โมนเทียม การให้คำปรึกษาทางพันธุกรรมทางการแพทย์เป็นสิ่งจำเป็นเกี่ยวกับความเป็นไปได้ของภาวะไทรอยด์ฮอร์โมนเทียมในลูกหลาน
การป้องกัน Pseudohypoparathyroidism:
คุณควรติดต่อแพทย์คนไหนหากคุณเป็นโรค Pseudohypoparathyroidism:
มีอะไรรบกวนคุณหรือเปล่า? คุณต้องการทราบข้อมูลโดยละเอียดเพิ่มเติมเกี่ยวกับ Pseudohypoparathyroidism สาเหตุ อาการ วิธีการรักษาและการป้องกัน ระยะของโรค และการรับประทานอาหารหลังจากนั้นหรือไม่ หรือต้องตรวจ? คุณสามารถ นัดหมายกับแพทย์– คลินิก ยูโรห้องปฏิบัติการพร้อมให้บริการคุณเสมอ! แพทย์ที่ดีที่สุดจะตรวจคุณและศึกษาคุณ สัญญาณภายนอกและจะช่วยคุณระบุโรคตามอาการ ให้คำแนะนำ ให้ความช่วยเหลือที่จำเป็น และทำการวินิจฉัย คุณก็ทำได้ โทรหาหมอที่บ้าน. คลินิก ยูโรห้องปฏิบัติการเปิดให้คุณตลอดเวลา
วิธีการติดต่อคลินิก:
หมายเลขโทรศัพท์ของคลินิกของเราในเคียฟ: (+38 044) 206-20-00 (หลายช่องทาง) เลขานุการคลินิกจะเลือกวันและเวลาที่สะดวกให้คุณมาพบแพทย์ พิกัดและทิศทางของเราระบุไว้ ดูรายละเอียดเพิ่มเติมเกี่ยวกับบริการทั้งหมดของคลินิก
(+38 044) 206-20-00
หากคุณเคยทำการวิจัยมาก่อน อย่าลืมนำผลไปพบแพทย์เพื่อขอคำปรึกษาหากไม่มีการศึกษา เราจะทำทุกอย่างที่จำเป็นในคลินิกของเราหรือกับเพื่อนร่วมงานในคลินิกอื่นๆ
คุณ? คุณจำเป็นต้องดูแลสุขภาพโดยรวมของคุณอย่างระมัดระวัง คนไม่ค่อยสนใจ. อาการของโรคและไม่รู้ว่าโรคเหล่านี้เป็นอันตรายถึงชีวิตได้ มีหลายโรคที่ในตอนแรกไม่ปรากฏในร่างกายของเรา แต่สุดท้ายกลับกลายเป็นว่าน่าเสียดายที่สายเกินไปที่จะรักษา แต่ละโรคมีอาการลักษณะเฉพาะของตัวเอง อาการภายนอก- เรียกว่า อาการของโรค. การระบุอาการเป็นขั้นตอนแรกในการวินิจฉัยโรคโดยทั่วไป ในการทำเช่นนี้คุณเพียงแค่ต้องทำปีละหลายครั้ง ได้รับการตรวจโดยแพทย์ที่ไม่เพียงแต่ป้องกันเท่านั้น โรคร้ายแต่ยังเพื่อรักษาสุขภาพจิตที่ดีทั้งในร่างกายและสิ่งมีชีวิตโดยรวม
หากคุณต้องการถามคำถามกับแพทย์ ให้ใช้ส่วนการให้คำปรึกษาออนไลน์ บางทีคุณอาจพบคำตอบสำหรับคำถามของคุณที่นั่นและอ่าน เคล็ดลับการดูแลตัวเอง. หากคุณสนใจรีวิวเกี่ยวกับคลินิกและแพทย์ ลองค้นหาข้อมูลที่คุณต้องการในส่วนนี้ ลงทะเบียนบนพอร์ทัลการแพทย์ด้วย ยูโรห้องปฏิบัติการเพื่อติดตามข่าวสารล่าสุด ข่าวล่าสุดและการอัปเดตข้อมูลบนเว็บไซต์ซึ่งจะถูกส่งถึงคุณทางอีเมลโดยอัตโนมัติ
โรคอื่นๆ ในกลุ่ม โรคระบบต่อมไร้ท่อ ภาวะโภชนาการผิดปกติ และความผิดปกติของระบบเผาผลาญ
| วิกฤต Addisonian (ภาวะต่อมหมวกไตไม่เพียงพอเฉียบพลัน) |
| มะเร็งต่อมน้ำเหลือง |
| Adiposogenital dystrophy (โรค Perchkranz-Babinski-Fröhlich) |
| กลุ่มอาการต่อมหมวกไต |
| อะโครเมกาลี |
| ความวิกลจริตทางโภชนาการ (โภชนาการเสื่อม) |
| อัลคาโลซิส |
| อัลแคปโตนูเรีย |
| อะไมลอยด์ซิส (อะไมลอยด์เสื่อม) |
| อะไมลอยโดซิสของกระเพาะอาหาร |
| อะไมลอยโดซิสในลำไส้ |
| อะไมลอยโดซิสเกาะตับอ่อน |
| อะไมลอยโดซิสในตับ |
| อะไมลอยโดซิสของหลอดอาหาร |
| ภาวะความเป็นกรด |
| ภาวะทุพโภชนาการโปรตีนและพลังงาน |
| โรคไอเซลล์ (mucolipidosis type II) |
| โรควิลสัน-โคโนวาลอฟ (โรคสมองเสื่อม) |
| โรคเกาเชอร์ (glucocerebroside lipidosis, glucocerebrosidosis) |
| โรคอิทเซนโก-คุชชิง |
| โรค Krabbe (เม็ดเลือดขาวเม็ดเลือดขาว) |
| โรคนีมันน์-พิค (sphingomyelinosis) |
| โรคฟาบรี้ |
| Gangliosidosis GM1 ประเภทที่ 1 |
| Gangliosidosis GM1 ประเภท II |
| Gangliosidosis GM1 ประเภทที่สาม |
| แกงลิโอซิโดซิส GM2 |
| Gangliosidosis GM2 ประเภทที่ 1 (ความโง่เขลาของ Tay-Sachs, โรค Tay-Sachs) |
| GM2 gangliosidosis type II (โรคของ Sandhoff, ความโง่เขลา amaurotic ของ Sandhoff) |
| Gangliosidosis GM2 ในเด็กและเยาวชน |
| ความใหญ่โต |
| ภาวะไขมันในเลือดสูง |
| Hyperaldosteronism รอง |
| hyperaldosteronism หลัก (กลุ่มอาการของ Conn) |
| ภาวะวิตามินเกิน D |
| ภาวะวิตามินเกิน A |
| ภาวะวิตามินเกิน E |
| ภาวะปริมาตรสูง |
| อาการโคม่าระดับน้ำตาลในเลือดสูง (เบาหวาน) |
| ภาวะโพแทสเซียมสูง |
| ภาวะแคลเซียมในเลือดสูง |
| ไขมันในเลือดสูงประเภทที่ 1 |
| ไขมันในเลือดสูงประเภท II |
| ไขมันในเลือดสูงประเภท III |
| ไขมันในเลือดสูงประเภท IV |
| ไขมันในเลือดสูงประเภท V |
| อาการโคม่าเกินขนาด |
| Hyperparathyroidism รอง |
| ภาวะต่อมพาราไทรอยด์ทำงานเกินปฐมภูมิ |
| Hyperplasia ของต่อมไทมัส (ต่อมไธมัส) |
| ภาวะโปรแลคติเนเมียสูง |
| ลูกอัณฑะทำงานผิดปกติ |
| ไขมันในเลือดสูง |
| ภาวะไขมันในเลือดต่ำ |
| อาการโคม่าฤทธิ์ลดน้ำตาลในเลือด |
| ภาวะ Hypogonadism |
| Hypogonadism ภาวะโปรแลกติเนมิกส์มากเกินไป |
| แยก Hypogonadism (ไม่ทราบสาเหตุ) |
| hypogonadism แต่กำเนิดปฐมภูมิ (อนาธิปไตย) |
| hypogonadism ที่ได้มาเบื้องต้น |
| ภาวะโพแทสเซียมต่ำ |
| ภาวะไทรอยด์ฮอร์โมนต่ำ |
| ภาวะ Hypopituitarism |
| ภาวะไทรอยด์ทำงานต่ำ |
| ไกลโคจีโนซิสประเภท 0 (aglycogenosis) |
| Glycogenosis ประเภทที่ 1 (โรคของ Gierke) |
| ไกลโคเจโนซิสประเภท II (โรคปอมเป) |
| ไกลโคจีโนซิสประเภทที่ 3 (โรคหัด, โรคฟอร์บส์, โรคเด็กซ์ทริโนซิสจำกัด) |
| ไกลโคจีโนซิสประเภทที่ 4 (โรคแอนเดอร์เซน, อะไมโลเพคติโนซิส, ไกลโคจีโนซิสแบบแพร่กระจายร่วมกับโรคตับแข็งในตับ) |
| Glycogenosis ประเภท IX (โรคของ Haga) |
| Glycogenosis ประเภท V (โรค McArdle, การขาด myophosphorylase) |
| Glycogenosis type VI (โรคของเธอ, ภาวะพร่องตับ) |
| Glycogenosis ประเภท VII (โรค Tarui, การขาด myophosphofructokinase) |
| Glycogenosis ประเภท VIII (โรคของทอมสัน) |
| ไกลโคจีโนซิสประเภท XI |
| ไกลโคเจโนซิสประเภท X |
| การขาด (ไม่เพียงพอ) ของวานาเดียม |
| การขาดแมกนีเซียม (ไม่เพียงพอ) |
| การขาดแมงกานีส (ไม่เพียงพอ) |
| การขาดทองแดง (ไม่เพียงพอ) |
| การขาด (ไม่เพียงพอ) ของโมลิบดีนัม |
| การขาด (ไม่เพียงพอ) ของโครเมียม |
| การขาดธาตุเหล็ก |
| การขาดแคลเซียม (การขาดแคลเซียมทางโภชนาการ) |
| การขาดธาตุสังกะสี (การขาดธาตุสังกะสีในอาหาร) |
| อาการโคม่า ketoacidotic เบาหวาน |
| ความผิดปกติของรังไข่ |
| คอพอกกระจาย (เฉพาะถิ่น) |
| วัยแรกรุ่นล่าช้า |
| เอสโตรเจนส่วนเกิน |
| การมีส่วนร่วมของต่อมน้ำนม |
| คนแคระ (เตี้ย) |
| ควาชิออร์กอร์ |
| โรคเต้านมอักเสบเรื้อรัง |
| แซนธินูเรีย |
| อาการโคม่ากรดแลคติค |
| Leucinosis (โรคน้ำเชื่อมเมเปิ้ล) |
| ไขมัน |
| ฟาร์เบอร์ ไลโปแกรนูโลมาโตซิส |
| Lipodystrophy (ความเสื่อมของไขมัน) |
| lipodystrophy ทั่วไป แต่กำเนิด (ซินโดรม Seyp-Lawrence) |
| ภาวะไขมันในเลือดสูงของกล้ามเนื้อ |
| lipodystrophy หลังการฉีด |
| lipodystrophy ปล้องก้าวหน้า |
| ไลโปมาโทซิส |
| Lipomatosis นั้นเจ็บปวด |
| มะเร็งเม็ดเลือดขาวชนิดเมตาโครมาติก |
| อาการโคม่า Myxedema |
Pseudohypoparathyroidism - เพียงพอแล้ว โรคที่หายากระบบโครงร่างซึ่งเป็นสาระสำคัญซึ่งเป็นการละเมิดการเผาผลาญแคลเซียมและฟอสฟอรัส ผู้ป่วยมักประสบกับความยับยั้งการพัฒนาจิตใจและร่างกาย โรคนี้เป็นกรรมพันธุ์
จากข้อเท็จจริงที่ว่าชื่อของโรคมีคำนำหน้าว่า "หลอก" เราสามารถเข้าใจได้ง่ายว่ามันเลียนแบบภาวะพาราไทรอยด์ฮอร์โมนต่ำ ชื่อที่สองของโรคนี้คือโรคกระดูกพรุนทางพันธุกรรมของอัลไบรท์ซึ่งตั้งชื่อตามแพทย์ผู้ศึกษาและบรรยายพยาธิสภาพนี้ในช่วงกลางศตวรรษที่ผ่านมา
ประเภทของ pseudohypoparathyroidism
Pseudohypoparathyroidism มีสองประเภท ขึ้นอยู่กับว่าระดับแคลเซียมในเลือดมีการเปลี่ยนแปลงหรือไม่ โรคอัลไบรท์ประเภทแรกก็เหมือนกัน อาการทางคลินิกเช่นเดียวกับในภาวะ hypoparathyroidism ที่ไม่ทราบสาเหตุ แต่มีลักษณะเฉพาะคือระดับแคลเซียมในเลือดลดลง ไม่มีความไวของเนื้อเยื่อต่อฮอร์โมนพาราไธรอยด์ ในทางกลับกัน pseudohypoparathyroidism ประเภทที่สองแตกต่างกันตรงที่ระดับแคลเซียมเป็นปกติ ดังนั้นประเภทนี้จึงเรียกว่า pseudopseudohypoparathyroidism อย่างไรก็ตามตามที่นักต่อมไร้ท่อระบุว่ารูปแบบหนึ่งสามารถเปลี่ยนเป็นอีกรูปแบบหนึ่งได้อย่างง่ายดายและสมาชิกในครอบครัวเดียวกันอาจมี ประเภทต่างๆโรคต่างๆ
สาเหตุของภาวะ pseudohypoparathyroidism
ความผิดปกติของการเผาผลาญฟอสฟอรัส - แคลเซียมเกิดขึ้นเนื่องจากการต้านทานเนื้อเยื่อต่อฮอร์โมนพาราไธรอยด์ซึ่งผลิตโดยต่อมพาราไธรอยด์ บนพื้นฐานของ pseudohypoparathyroidism ปรากฏการณ์ของความไวของเนื้อเยื่อบกพร่องต่อฮอร์โมนที่ผลิตโดยต่อมไร้ท่อหรือภายนอกได้รับการยืนยันเป็นครั้งแรก
Pseudohypoparathyroidism เป็นพยาธิสภาพทางพันธุกรรม มีสาเหตุมาจากกลุ่มอาการที่มีมา แต่กำเนิด - ตัวรับเซลล์จำเพาะ - ฮอร์โมนพาราไธรอยด์ - อะดีนิเลตไซคลอสซึ่งเป็นสาเหตุที่เนื้อเยื่อส่วนปลายสูญเสียความไวต่อฮอร์โมนพาราไธรอยด์
ใครที่เสี่ยงต่อการเกิด pseudohypoparathyroidism มากที่สุด? ประการแรกคือลูกของผู้ป่วยและญาติคนอื่น ๆ เนื่องจากโรคนี้เป็นกรรมพันธุ์และมีออโตโซมที่โดดเด่น ผู้หญิงต้องทนทุกข์ทรมานจากโรคอัลไบรท์บ่อยขึ้น ผู้เชี่ยวชาญอธิบายเรื่องนี้โดยข้อเท็จจริงที่ว่าผู้ชายที่มีภาวะ pseudohypoparathyroidism มีอัตราการเจริญพันธุ์ต่ำ และการถ่ายทอดทางพันธุกรรมนั้นถ่ายทอดจากพ่อสู่ลูกเฉพาะในบางกรณีเท่านั้น
ไม่มีสถิติว่าโรคนี้พบได้บ่อยเพียงใดมีการอธิบายผู้ป่วยประมาณสามร้อยรายในงานทางการแพทย์
อาการของ pseudohypoparathyroidism
- ปัญหาเกี่ยวกับอวัยวะ ระบบต่อมไร้ท่อด้วย pseudohypoparathyroidism อาการหลักจะถูกกำหนดด้วย: ผู้ป่วยมักจะแข็งแรง, เตี้ยกว่าคนอื่น, เนื่องจากสั้นลง แขนขาส่วนล่าง, เป็นโรคอ้วน, ระดับน้ำตาลในเลือดสูงขึ้น, ใบหน้าเป็นรูปพระจันทร์;
- ตัวแทนของเพศที่ยุติธรรมกว่าได้รับผลกระทบ รอบประจำเดือน. บ่อยครั้งที่สัญญาณเหล่านี้มาพร้อมกับ acromegaly, gynecomastia, กระบวนการแพ้ภูมิตัวเองต่างๆ, กลุ่มอาการ Itsenko-Cushing และโรคอื่น ๆ ในผู้ป่วยดังกล่าวความเสี่ยงของโรคเบาหวานและโรคเบาจืดก็สูงขึ้นเช่นกัน
- ผู้ที่เป็นโรคอัลไบรท์มักจะบ่นว่ามีปัญหากับฟัน (เคลือบฟันเสียหาย) ผู้ป่วยยังถูกรบกวนด้วยการอาเจียน การชักแบบโทนิค - ทั้งที่เกิดขึ้นเองและเกิดขึ้นภายใต้อิทธิพลของสารระคายเคือง อาจสังเกตปัสสาวะในเลือดได้ มีความเสี่ยงที่จะเป็นต้อกระจก มีลักษณะเป็นภาวะปัญญาอ่อน เด็กที่เป็นโรคนี้ไม่สามารถรับมือกับหลักสูตรของโรงเรียนได้ ความจำลดลง
- ใน ระบบข้อเข่าเสื่อมการเปลี่ยนแปลงที่สำคัญก็เกิดขึ้นเช่นกัน - โรคกระดูกพรุนกระจายปรากฏขึ้น, ซีสต์ (เนื้องอกสีน้ำตาล) พัฒนาขึ้น คนไข้ที่เป็นโรค pseudohypoparathyroidism มีแนวโน้มที่จะกระดูกหักหรือความผิดปกติของกระดูกมากกว่า กระดูกฝ่ามือชิ้นที่ 1, 4 และ 5 และกระดูกฝ่าเท้าจะสั้นลง และสังเกตสัญญาณของการสลายของกระดูกที่มือและเท้า นอกจากนี้แคลเซียมซึ่งถูกปล่อยออกมาจากกระดูกเริ่มสะสมในเนื้อเยื่อใต้ผิวหนังทำให้เกิดการกลายเป็นปูน (สิ่งนี้เกิดขึ้นกับ myositis ossificans) ในบริเวณข้อต่อและโพรงกะโหลก การกลายเป็นปูนยังมีอยู่ในกล้ามเนื้อ ไต กล้ามเนื้อหัวใจตาย และบนผนังหลอดเลือดแดงใหญ่
- อื่น จุดเด่นโรคอัลไบรท์ - ผิวคล้ำ จุดสีน้ำตาลกระจายไปทั่วร่างกาย (ทั้งด้านหนึ่งและอีกด้านหนึ่ง)
ส่วนความสามารถในการทำงานนั้นขึ้นอยู่กับความเข้มงวดของกระบวนการและประสิทธิภาพ การรักษาด้วยยา. หากรูปแบบของโรคแฝงอยู่และไม่มีการโจมตีที่ชัดเจน ความสามารถในการทำงานจะถูกรักษาไว้บางส่วน แต่มีข้อ จำกัด หลายประการ - โดยเฉพาะอย่างยิ่งคุณไม่สามารถทำงานกับกลไกการเคลื่อนไหวหรือในการขนส่งได้ ห้ามใช้ความเครียดทางระบบประสาทและการใช้แรงงานมากเกินไป หากมีการชักบ่อยครั้ง มีการแสดงอาการปัญญาอ่อนอย่างชัดเจน หรือมีความบกพร่องทางการมองเห็นอย่างมากเนื่องจากต้อกระจก ผู้ป่วยดังกล่าวจะถือว่าพิการแล้ว
การวินิจฉัยโรค pseudohypoparathyroidism
อาการจะค่อนข้างเฉพาะเจาะจง และหากเริ่มปรากฏ ควรติดต่อแพทย์ผู้เชี่ยวชาญทันที เป็นช่วงต้นและ การวินิจฉัยที่ถูกต้องช่วยให้มั่นใจถึงความสำเร็จของการรักษา
เนื่องจากโรคออลไบรท์เป็นโรคที่มีมา แต่กำเนิด จึงมักได้รับการวินิจฉัยกับผู้ป่วยในวัยเด็ก (ก่อนวัยเรียน, จูเนียร์ วัยเรียน). แพทย์ทางพันธุกรรมให้ข้อสรุปตามอาการทางคลินิก นอกจากนี้ยังใช้วิธีการตรวจทางห้องปฏิบัติการและเครื่องมือด้วย
โดยเฉพาะอย่างยิ่ง เพื่อชี้แจงการวินิจฉัย ผู้ป่วยบริจาคเลือด (ในคนไข้ที่เป็น pseudohypoparathyroidism ระดับแคลเซียมต่ำ และฟอสฟอรัสและฮอร์โมนพาราไธรอยด์สูง กิจกรรมอัลคาไลน์ฟอสฟาเตสในเลือดสูง) ปัสสาวะ (ในผู้ป่วยนี้ การวิเคราะห์จะแสดงฟอสฟอรัสและแคลเซียมที่ถูกขับออกมาลดลง) มีการทดสอบพิเศษเพื่อแสดงให้เห็นว่าเนื้อเยื่อท่อไตของผู้ป่วยมีความไวต่อฮอร์โมนพาราไธรอยด์เพียงใด บางครั้งจำเป็นต้องประเมินระดับของไฮดรอกซีโพรลีน
วิธีการใช้เครื่องมือเกี่ยวข้องกับการวินิจฉัยด้วยรังสีเอกซ์ (จะให้ข้อมูลได้ดีที่สุดในการระบุการเปลี่ยนแปลงเฉพาะในกระดูกและเนื้อเยื่อของกล้ามเนื้อ)
การรักษาภาวะ pseudohypoparathyroidism
พยาธิวิทยาทางพันธุกรรมที่หายากนี้ได้รับการรักษาด้วยการเสริมแคลเซียม แพทย์เลือกขนาดยาตามปริมาณแคลเซียมที่จำเป็นในการรักษาสภาวะสมดุล และเพื่อให้แคลเซียมดูดซึมได้โดยไม่มีปัญหาจึงรวมการเตรียมวิตามินดีไว้ในแผนการรักษาด้วย นอกจากนี้ แพทย์สำหรับผู้ป่วยที่มีภาวะ pseudohypoparathyroidism จะพัฒนาอาหารที่ปริมาณอาหารที่มีฟลูออไรด์จะถูกจำกัด อาหารนี้จะช่วยปรับความเข้มข้นของแคลเซียมในเลือดให้เป็นปกติและกำจัดสัญญาณของภาวะพาราไทรอยด์ฮอร์โมนทุติยภูมิทุติยภูมิ
ถ้าโรคออลไบร์ทเกิดขึ้นร่วมด้วย ความผิดปกติของต่อมไร้ท่อแล้วอาจจำเป็นต้องบำบัดด้วยฮอร์โมนทดแทน อาการชักจะบรรเทาลงด้วยสารละลายแคลเซียมคลอไรด์หรือแคลเซียมกลูโคเนตซึ่งฉีดเข้าเส้นเลือดดำ หากต้อกระจกปรากฏขึ้น จักษุแพทย์จะรักษาและนักจิตวิทยาจะช่วยบรรเทาความบกพร่องทางสติปัญญา
ด้วยสูตรการรักษาที่คิดมาอย่างดี การพยากรณ์โรคจึงเป็นที่น่าพอใจมาก แต่ผู้ที่เป็นโรค pseudohypoparathyroidism ควรปรึกษานักพันธุศาสตร์เป็นประจำเพื่อป้องกันโรคในลูกหลาน
การป้องกันการเกิด pseudohypoparathyroidism
พิจารณาว่าโรคนี้เป็นกรรมพันธุ์แล้ว วิธีเดียวเท่านั้นการป้องกันสำหรับผู้ที่มีประวัติครอบครัวเป็นโรค pseudohypoparathyroidism คือการไปพบนักพันธุศาสตร์ทางการแพทย์เพื่อขอคำปรึกษาเป็นประจำ
(โรคอัลไบรท์) เป็นภาวะกระดูกเสื่อมทางพันธุกรรมที่เกิดจากความต้านทานของเนื้อเยื่อส่วนปลายต่อฮอร์โมนพาราไธรอยด์ซึ่งมาพร้อมกับความผิดปกติของการเผาผลาญแคลเซียมฟอสฟอรัสทำให้การพัฒนาทางร่างกายและจิตใจล่าช้า Pseudohypoparathyroidism เกิดขึ้นพร้อมกับอาการของโรคกระดูกพรุนแบบกระจาย การชักแบบโทนิค การแตกหักและการเสียรูปของกระดูก การสะสมแคลเซียมในกล้ามเนื้อและหลอดเลือด การก่อตัวของนิ่วในทางเดินปัสสาวะ การชะลอการเจริญเติบโต และภาวะปัญญาอ่อน ในการวินิจฉัย pseudohypoparathyroidism จะกำหนดระดับแคลเซียม, อัลคาไลน์ฟอสฟาเตสและฮอร์โมนพาราไธรอยด์ในเลือด การขับถ่ายฟอสฟอรัสและแคลเซียมในปัสสาวะ การทดสอบการทำงานด้วยการแนะนำฮอร์โมนพาราไธรอยด์ การวินิจฉัยด้วยรังสีเอกซ์ การรักษา pseudohypoparathyroidism เกี่ยวข้องกับการแก้ไขภาวะน้ำตาลในเลือดต่ำ
ไอซีดี-10
E20.1

ข้อมูลทั่วไป
Pseudohypoparathyroidism เป็นพยาธิสภาพทางพันธุกรรมที่หาได้ยาก ในด้านต่อมไร้ท่อมีเพียงประมาณ 300 รายเท่านั้นที่ทราบโรคนี้ Pseudohypoparathyroidism ในอาการของมันคล้ายกับ hypoparathyroidism อย่างไรก็ตามหากภาวะ hypoparathyroidism ขึ้นอยู่กับการขาดฮอร์โมนพาราไธรอยด์ขั้นต้นซึ่งเกิดจากกิจกรรมการทำงานของต่อมพาราไธรอยด์ลดลงดังนั้นในโรคของ Albright กลุ่มอาการ pseudohypoparathyroid นั้นมีสาเหตุมาจากการละเมิดความไวของเนื้อเยื่อเป้าหมายต่อการทำงานของพาราไธรอยด์ ฮอร์โมนที่มีระดับการหลั่งเพียงพอ
Pseudohypoparathyroidism ได้รับการอธิบายครั้งแรกในปี 1942 F. Albright และตามที่ผู้เขียนกล่าวไว้ มันถูกเรียกว่า "โรคของ Albright" หรือ "โรคกระดูกเสื่อมทางพันธุกรรมของ Albright" ตัวแปรแรกของโรคคือลักษณะความผิดปกติในการพัฒนาโครงกระดูก, ความต้านทานของเนื้อเยื่อส่วนปลายต่อฮอร์โมนพาราไธรอยด์, ภาวะแคลเซียมในเลือดต่ำและ ภาพทางคลินิกคล้ายกับภาวะพาราไทรอยด์ฮอร์โมนต่ำที่ไม่ทราบสาเหตุ ตัวแปรที่สองของโรคอัลไบรท์เกิดขึ้นตามประเภทของ normocalcemic และเรียกว่า pseudopseudohypoparathyroidism ในวรรณคดี มีการอธิบายกรณีของการพัฒนาในครอบครัวหนึ่งของโรคทั้งแบบปกติและแบบ hypocalcemic ตลอดจนการเปลี่ยนรูปแบบหนึ่งไปสู่อีกรูปแบบหนึ่ง
สาเหตุของภาวะ pseudohypoparathyroidism
ในรูปแบบต่างๆ ของ pseudohypoparathyroidism โรคนี้มีลักษณะทางพันธุกรรมโดยมีการถ่ายทอดทางพันธุกรรมแบบออโตโซมที่เชื่อมโยงกับโครโมโซม X การศึกษาสายเลือดพบว่าจำนวนผู้หญิงที่มีภาวะ pseudohypoparathyroidism สูงกว่าจำนวนผู้ชายที่ป่วยถึง 2 เท่า นอกจากนี้โรคอัลไบรท์ไม่ได้แพร่เชื้อจากพ่อสู่ลูก
Pseudohypoparathyroidism เกิดจากการต้านทานทางพันธุกรรมของโครงกระดูกและไตต่อการทำงานของฮอร์โมนพาราไธรอยด์ นี่เป็นเพราะข้อบกพร่องในตัวรับจำเพาะของพลาสมาเมมเบรนของเซลล์เป้าหมายและการขาดเอนไซม์ adenylate cyclase, โปรตีนไคเนส, cyclic 3, 5-adenosine monophosphate (AMP) ใน tubules ส่วนปลายของ nephron ซึ่งมาพร้อมกับ การดูดซึมฟอสฟอรัสอีกครั้ง การขับถ่ายฟอสฟอรัสในปัสสาวะลดลงทำให้เกิดภาวะฟอสเฟตในเลือดสูงและภาวะแคลเซียมในเลือดต่ำทุติยภูมิ
เนื่องจากใน pseudohypoparathyroidism ต่อมพาราไธรอยด์ยังคงไม่บุบสลาย ภาวะแคลเซียมในเลือดต่ำอาจทำให้เกิดการกระตุ้นการหลั่งฮอร์โมนพาราไธรอยด์และการพัฒนาของภาวะพาราไทรอยด์ฮอร์โมนทุติยภูมิทุติยภูมิ ด้วย pseudohypoparathyroidism มักจะมีการชดเชย hyperplasia ของต่อมพาราไธรอยด์ (การพัฒนาของ adenomas ไม่ปกติ) การเปลี่ยนแปลงของเนื้อเยื่อกระดูก (โรคกระดูกพรุน, ซีสต์), การสะสมของแคลเซียมใน กล้ามเนื้อโครงร่าง,เนื้อเยื่อใต้ผิวหนังตลอดจนในไต, ผนังหลอดเลือด, กล้ามเนื้อหัวใจตาย, เยื่อบุตาและกระจกตา Pseudohypoparathyroidism มักใช้ร่วมกับความดันโลหิตสูง เบาหวาน หลอดเลือดแดงอักเสบ และโรคข้ออักเสบหลายส่วน
อาการของ pseudohypoparathyroidism
อาการทางคลินิกของ pseudohypoparathyroidism มีความคล้ายคลึงกับอาการของ hypoparathyroidism ที่ไม่ทราบสาเหตุ อาการทางต่อมไร้ท่อของภาวะไทรอยด์ฮอร์โมนเทียม ได้แก่ รูปร่างเตี้ย โรคอ้วนและน้ำตาลในเลือดสูง หน้าพระจันทร์ และประจำเดือนน้อย โรคของออลไบรท์มักรวมกับความผิดปกติของต่อมไร้ท่ออื่น ๆ - อะโครเมกาลี, กลุ่มอาการคุชชิง, gynecomastia, พร่องหรือต่อมไทรอยด์ทำงานเกิน
อาการทางข้อกระดูกของ pseudohypoparathyroidism ได้แก่ brachydactyly โดยที่ metacarpals และ metacarpals ที่ 1, 4 และ 5 สั้นลงอย่างเห็นได้ชัด; exostoses, dyschondroplasia, การเปลี่ยนแปลงในปลาย epiphyseal ของกระดูก, การสลายของกระดูกของนิ้วมือ โดดเด่นด้วยการงอกของฟันล่าช้า, hypoplasia ของเคลือบฟัน, ขบวนการสร้างกระดูกใต้ผิวหนัง
อาการทางระบบประสาทของ pseudohypoparathyroidism จะแสดงโดยการโจมตีของอาการชักแบบโทนิคซึ่งอาจเกิดขึ้นได้เองหรือภายใต้อิทธิพลของสารระคายเคือง สำหรับผู้ป่วยที่มีภาวะ pseudohypoparathyroidism ภาวะปัญญาอ่อนเป็นเรื่องปกติ อาการอื่น ๆ ของ pseudohypoparathyroidism ได้แก่ urolithiasis, ปัสสาวะเป็นเลือด, อาเจียนและการพัฒนาของต้อกระจกแม่และเด็ก ด้วย pseudopseudohypoparathyroidism ไม่มีภาวะน้ำตาลในเลือดต่ำ, ภาวะฟอสเฟตในเลือดสูง, โรคกระดูกพรุนและอาการชัก
การวินิจฉัยโรค pseudohypoparathyroidism
ในกรณีทั่วไป pseudohypoparathyroidism ได้รับการวินิจฉัยในเด็กอายุ 5-10 ปีโดยคำนึงถึงอาการลักษณะห้องปฏิบัติการและข้อมูลทางรังสีวิทยา ในคนไข้ที่เป็นโรค pseudohypoparathyroidism, ภาวะน้ำตาลในเลือดต่ำ, ภาวะฟอสเฟตในเลือดสูง, กิจกรรมปกติหรือเพิ่มขึ้นของอัลคาไลน์ฟอสฟาเตสในเลือดและระดับฮอร์โมนพาราไธรอยด์ที่เพิ่มขึ้นในเลือดจะถูกกำหนด การขับแคลเซียมและฟอสฟอรัสในปัสสาวะลดลง
ความไม่รู้สึกของท่อไตต่อฮอร์โมนพาราไธรอยด์ได้รับการยืนยันโดยการทดสอบโดยพิจารณาจากปริมาณฟอสเฟตและอะดีโนซีนโมโนฟอสเฟตแบบไซคลิกที่ถูกขับออกมาในปัสสาวะ ใน pseudohypoparathyroidism เพื่อตอบสนอง การบริหารทางหลอดเลือดดำฮอร์โมนพาราไธรอยด์ ไม่มีปริมาณฟอสเฟตและแคมป์ในปัสสาวะเพิ่มขึ้นอย่างมีนัยสำคัญ
ในกรณีของ pseudohypoparathyroidism จำเป็นต้องมีการแก้ไขภาวะน้ำตาลในเลือดต่ำดังนั้นการบำบัดด้วยการเตรียมแคลเซียมจึงดำเนินการในปริมาณที่ช่วยให้รักษาความเข้มข้นของแคลเซียมตามปกติในเลือด ปริมาณวิตามินดีและรูปแบบที่ออกฤทธิ์จะแสดงภายใต้การควบคุมความเข้มข้นของแคลเซียมในเลือด การรักษาด้วยฮอร์โมนพาราไธรอยด์ไม่ได้ผล เพื่อทำให้ความเข้มข้นของแคลเซียมเป็นปกติและกำจัดอาการของภาวะพาราไธรอยด์เกินขั้นทุติยภูมิจำเป็นต้องรับประทานอาหารที่มีปริมาณฟอสฟอรัสจำกัด
ในกรณีที่การทำงานของต่อมอื่นไม่เพียงพอจะทำการบำบัดทดแทนด้วยฮอร์โมนที่เหมาะสม หากเกิดอาการชัก ให้ระบุการให้สารละลายแคลเซียมทางหลอดเลือดดำ
การวินิจฉัยและการรักษาอย่างมีเหตุผลของ pseudohypoparathyroidism อย่างทันท่วงทีช่วยให้เราสามารถพูดคุยเกี่ยวกับการพยากรณ์โรคเชิงบวกต่อชีวิตและความเป็นไปได้ในการควบคุมการดำเนินโรค เมื่อพิจารณาถึงลักษณะทางพันธุกรรมของ pseudohypoparathyroidism แนะนำให้ทำการให้คำปรึกษาทางพันธุกรรมทางการแพทย์เพื่อประเมินความเสี่ยงของโรคอัลไบรท์ในลูกหลาน
![]()
คำอธิบาย:
Pseudohypoparathyroidism (กรีก pseudēs false +; คำพ้องความหมาย: Osteodystrophy ทางพันธุกรรมของ Albright, โรคของ Albright) เป็นโรคทางพันธุกรรมที่หายากของระบบโครงกระดูก, จำลองภาวะ hypoparathyroidism และโดดเด่นด้วยการเผาผลาญแคลเซียมและฟอสฟอรัสบกพร่อง; มักมาพร้อมกับพัฒนาการทางจิตใจและร่างกายที่ล่าช้า
อาการ:
อาการทางคลินิกของ pseudohypoparathyroidism มีความคล้ายคลึงกับอาการของ hypoparathyroidism ที่ไม่ทราบสาเหตุ มีการโจมตีแบบโทนิคที่เกิดขึ้นเองตามธรรมชาติหรืออยู่ภายใต้อิทธิพลของการระคายเคือง การกลายเป็นปูนในเนื้อเยื่อใต้ผิวหนังมีแนวโน้มที่จะเป็นแผล ขบวนการสร้างกระดูกใต้ผิวหนังมักรุนแรงถึงขั้นเลียนแบบการสร้างกระดูก มีลักษณะพิเศษคือ ปัญญาอ่อน การเจริญเติบโตช้า ใบหน้ารูปพระจันทร์ และโดยเฉพาะอย่างยิ่งกระดูกฝ่ามือชิ้นที่ 1, 4 และ 5 และกระดูกฝ่าเท้าสั้นลง อาจมีการสังเกต exostoses หลายครั้ง dyschondroplasia และอาการทุติยภูมิในรูปแบบของการสลายกระดูกของนิ้วใต้ผิวหนัง การเปลี่ยนแปลงใน epiphyses ของกระดูกจะเหมือนกับใน fibrous Osteodysplasia มักสังเกตเห็นการอาเจียนเช่นเดียวกับปัสสาวะเนื่องจากการก่อตัวของนิ่วออกซาเลตในทางเดินปัสสาวะ, ตรวจพบต้อกระจกแม่และเด็ก, และ hypoplasia ของเคลือบฟัน
ในคนไข้ที่เป็น pseudohypoparathyroidism พร้อมกับความไวของอวัยวะเป้าหมายต่อฮอร์โมนพาราไธรอยด์ลดลง อาจสังเกตความต้านทานต่อฮอร์โมนอื่น ๆ ที่ขึ้นอยู่กับระบบ adenylate cyclase เช่นอวัยวะสืบพันธุ์ต่อฮอร์โมน gonadotropic ต่อมไทรอยด์ต่อฮอร์โมนกระตุ้นต่อมไทรอยด์ , อวัยวะเป้าหมายไปที่ฮอร์โมนกลูคากอนและแอนติไดยูเรติก มีอุบัติการณ์ของโรคแพ้ภูมิตัวเองและเบาหวานเพิ่มขึ้นและมีการสังเกตพบ
นอกจากนี้ยังมี pseudopseudohypoparathyroidism ซึ่งมีลักษณะเฉพาะโดยไม่มีอาการชักและโรคกระดูกพรุน
สาเหตุ:
สาเหตุของ pseudohypoparathyroidism เป็นข้อบกพร่องที่มีมา แต่กำเนิด - ความไม่รู้สึกของเนื้อเยื่อส่วนปลายต่อการกระทำของ PTH
การรักษา:
สำหรับการรักษามีการกำหนดดังต่อไปนี้:
การรักษาภาวะแคลเซียมในเลือดต่ำประกอบด้วยการสั่งจ่ายแคลเซียมเสริมในปริมาณที่เพียงพอเพื่อรักษาความเข้มข้นของแคลเซียมในเลือดให้เป็นปกติ การบำบัดด้วยวิตามินดีมีความสำคัญอย่างยิ่ง ขนาดยาเริ่มแรกคำนวณจาก 2,000 IU/น้ำหนักตัวกก. ต่อวัน แต่ไม่เกิน 100,000 IU ต่อวัน เพื่อหลีกเลี่ยงการเตรียมวิตามินดีเกินขนาดจำเป็นต้องตรวจสอบความเข้มข้นของแคลเซียมในเลือดทุก 3-7 วันในช่วงสองสัปดาห์แรกของการรักษาและทุกเดือนในช่วง 2-3 เดือนข้างหน้า เมื่อความเข้มข้นของแคลเซียมในเลือดถึงระดับคงที่แล้ว ก็เพียงพอที่จะตรวจดูทุกๆ 2-3 เดือน คุณสามารถใช้แคลซิทริน, ไดไฮโดรทาคิสเตอรอล, ออกไซด์วิตรวมถึงการเตรียมวิตามินดีในรูปแบบอื่น ๆ การรับประทานอาหารที่มีข้อ จำกัด ของฟอสฟอรัสจะช่วยให้ความเข้มข้นของแคลเซียมในเลือดเป็นปกติและกำจัดอาการของภาวะพาราไทรอยด์ฮอร์โมนทุติยภูมิทุติยภูมิ ในกรณีที่ต่อมไร้ท่ออื่นไม่เพียงพอจะทำการบำบัดทดแทนด้วยฮอร์โมนที่เหมาะสม การรักษาด้วยฮอร์โมนพาราไธรอยด์ไม่ได้ผล เพื่อหยุดการโจมตีแบบชักจะมีการให้สารละลายแคลเซียมคลอไรด์หรือแคลเซียมกลูโคเนต 10% ทางหลอดเลือดดำ ทางปาก - สารละลายแคลเซียมคลอไรด์ 5-10%, 1 ช้อนโต๊ะ 3-4 ครั้งต่อวัน: แคลเซียมกลูโคเนต, แคลเซียมแลคเตต - มากถึง 10 กรัมต่อวัน
การพยากรณ์โรคด้วยการบำบัดอย่างมีเหตุผลเป็นสิ่งที่ดี เมื่อพิจารณาถึงลักษณะทางพันธุกรรมของภาวะไทรอยด์ฮอร์โมนเทียม การให้คำปรึกษาทางพันธุกรรมทางการแพทย์เป็นสิ่งจำเป็นเกี่ยวกับความเป็นไปได้ของภาวะไทรอยด์ฮอร์โมนเทียมในลูกหลาน
อี.วี. Tozliyan แพทย์ต่อมไร้ท่อในเด็ก นักพันธุศาสตร์ Ph.D., I.V. Shulyakova นักประสาทวิทยา Ph.D.
โดดเดี่ยว การแบ่งส่วนโครงสร้าง"สถาบันวิจัยคลินิกกุมารเวชศาสตร์" สถาบันการศึกษางบประมาณของรัฐสำหรับการศึกษาวิชาชีพชั้นสูง "มหาวิทยาลัยการแพทย์วิจัยแห่งชาติรัสเซียตั้งชื่อตาม N.I. Pirogov" กระทรวงสาธารณสุขแห่งสหพันธรัฐรัสเซีย กรุงมอสโก
คำหลัก:
เด็ก, pseudohypoparathyroidism, โรคกระดูกพรุนทางพันธุกรรมของ Albright, โรคอ้วน, ภาวะแคลเซียมในเลือดต่ำ, การวินิจฉัย, การดื้อต่อฮอร์โมนพาราไธรอยด์
คำสำคัญ:เด็ก, pseudohypoparathyroidism, โรคกระดูกพรุนทางพันธุกรรมของอัลไบรท์, โรคอ้วน, ภาวะแคลเซียมในเลือดต่ำ, การวินิจฉัย, การดื้อต่อฮอร์โมนพาราไธรอยด์
Pseudohypoparathyroidism (กรีก pseudes - false + hypoparathyroidism; คำพ้องความหมาย: Osteodystrophy ทางพันธุกรรมของ Albright, โรคไก่ชวา) เป็นโรคทางพันธุกรรมที่หายากของระบบโครงร่าง, จำลอง hypoparathyroidism และโดดเด่นด้วยการเผาผลาญแคลเซียมและฟอสฟอรัสบกพร่อง; มักมาพร้อมกับพัฒนาการทางจิตใจและร่างกายที่ล่าช้า โรคนี้ได้รับการอธิบายครั้งแรกโดยนักต่อมไร้ท่อชาวอเมริกัน Albright F. ในปี 1942 ความชุกของโรคอยู่ที่ 7.9 ต่อ 1 ล้านคน
ข้อมูลทางพันธุกรรม
Pseudohypoparathyroidism (PHP) เป็นโรคที่มีความหลากหลายทางพันธุกรรม ข้อมูลประเภทของการถ่ายทอดทางพันธุกรรมนั้นขัดแย้งกัน: ทั้งประเภท X-linked dominant และ autosomal dominant, autosomal recessive ในกรณีส่วนใหญ่ การพัฒนาของกระดูกเสื่อมทางพันธุกรรมของอัลไบรท์นั้นสัมพันธ์กับการกลายพันธุ์ในตำแหน่ง 20q13 ของยีน GNAS1 ที่อยู่บนโครโมโซม 20 (Patten et al., 1990) ซึ่งเข้ารหัสโปรตีน Gs-alpha ที่เกี่ยวข้องกับตัวรับฮอร์โมนพาราไธรอยด์ (PTH) . นอกจากนี้ ยังตรวจพบฟีโนไทป์ที่คล้ายกันในผู้ป่วยที่มีการลบคั่นระหว่างแขนยาวของโครโมโซม 2 ที่ตำแหน่ง 2q37
การเกิดโรค
การเกิดโรคของ pseudohypoparathyroidism นั้นขึ้นอยู่กับการต้านทานทางพันธุกรรมของไตและโครงกระดูกต่อการทำงานของฮอร์โมนพาราไธรอยด์อันเป็นผลมาจากข้อบกพร่องในคอมเพล็กซ์ "ตัวรับไซโตรีเซพเตอร์ - ฮอร์โมนพาราไธรอยด์ - อะดีนีเลตไซคลอส" ที่ซับซ้อนซึ่งขัดขวางการก่อตัวของไซคลิก 3"- , 5"-อะดีโนซีนโมโนฟอสเฟต (แคมป์) ในไตซึ่งเป็นสื่อกลางในเซลล์ของการทำงานของฮอร์โมนพาราไธรอยด์ต่อกระบวนการเผาผลาญ Pseudohypoparathyroidism เกิดจากพันธุกรรม โรคต่างกันในผู้ป่วยบางราย ตัวรับไซโตรีเซพเตอร์เองที่จับกับฮอร์โมนพาราไธรอยด์มีข้อบกพร่อง (pseudohypoparathyroidism ประเภท 1A) ส่วนตัวอื่นๆ มีข้อบกพร่องในโปรตีนที่จับกับนิวคลีโอไทด์ ซึ่งอยู่ในชั้นไขมันสองชั้นของเยื่อหุ้มเซลล์และทำหน้าที่จับตัวรับกับอะดีนิเลตไซคเลส (ประเภท 1B pseudohypoparathyroidism) ผู้ป่วยบางรายมีภาวะขาดเอนไซม์ adenylate cyclase เอง (pseudohypoparathyroidism ประเภท 2) การขาดแคมป์ที่เกิดจากข้อบกพร่องเหล่านี้นำไปสู่การหยุดชะงักของการสังเคราะห์โปรตีนเฉพาะที่กำหนดผลกระทบทางชีวภาพของฮอร์โมนพาราไธรอยด์ ดังนั้นความไวของอวัยวะเป้าหมายต่อฮอร์โมนพาราไธรอยด์จึงหายไป
ลักษณะทางคลินิก
ปัจจุบันมี 4 ตัว รูปแบบทางคลินิกพยาธิสภาพ: ประเภท 1A, 1B, 1C และ 2 ความรู้เกี่ยวกับลักษณะและข้อมูลทางคลินิกและทางชีวเคมี การวิจัยทางพันธุกรรมช่วยให้สามารถวินิจฉัยแยกโรคได้ภายในรูปแบบทาง nosological
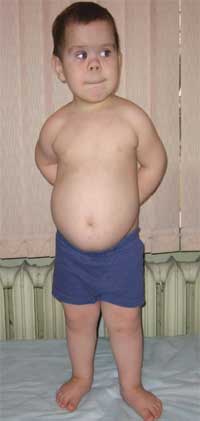
สัญญาณทั่วไปที่ทำให้สงสัยว่าโรคนี้คือความไม่สมส่วนในการพัฒนาทางกายภาพ รูปร่างเตี้ย (จนแคระแกร็น) เนื่องจากแขนขาส่วนล่างสั้นลง (ภาพที่ 1) brachydactyly (ภาพที่ 2) และ "รูปดวงจันทร์" ทรงกลม ใบหน้า (ภาพที่ 3) บางครั้งมีการสังเกต exostoses และ aplasia ของฟัน
รูปภาพที่ 1
รูปร่างเด็กที่มีภาวะกระดูกเสื่อมของอัลไบรท์
(ลักษณะของฟีโนไทป์ ความสูงสั้น เนื่องจากแขนขาส่วนล่างสั้นลง)
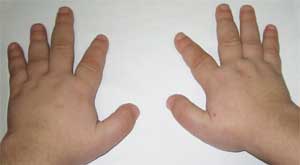
รูปภาพที่ 2
ลักษณะของระบบโครงกระดูกของผู้ป่วย
ด้วยโรคกระดูกเสื่อมของอัลไบรท์
(brachydactyly - นิ้วสั้นลง)
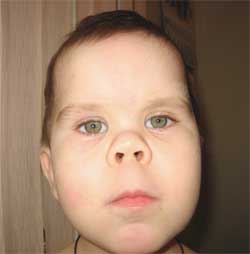
รูปภาพที่ 3
คุณสมบัติของฟีโนไทป์ของเด็ก
ด้วยโรคกระดูกเสื่อมของอัลไบรท์
(หน้ากลมรูปพระจันทร์)
สัญญาณที่ทำให้เกิดโรคคือการทำให้กระดูกฝ่ามือและกระดูกฝ่าเท้า I, III และ V สั้นลง (โดยเฉพาะ III และ IV) ซึ่งเป็นผลมาจากการที่นิ้ว II บนมือและเท้ายาวกว่านิ้วอื่น ๆ และเมื่อมือกำแน่น เป็นกำปั้นไม่มีส่วนนูนในบริเวณของข้อต่อ metacarpophalangeal IV และ V - ที่เรียกว่า brachymetaphalangism นอกจากนี้ยังตรวจพบช่วงกว้างที่สั้น กะโหลกศีรษะหนาขึ้น และกระดูกไม่มีแร่ธาตุ (โรคกระดูกพรุน) และโรคอ้วนอีกด้วย
ปัญญาอ่อน(มักมีความรุนแรงปานกลาง) พบได้ในผู้ป่วยประมาณ 20% ตามที่ผู้เขียนบางคนระบุว่า ภาวะปัญญาอ่อนเกิดขึ้นใน 70% ของกรณีที่มีภาวะน้ำตาลในเลือดต่ำและใน 30% ของกรณีในภาวะน้ำตาลในเลือดปกติ กระบวนการทางจิตในผู้ป่วยจะชะลอตัวลง ในสถานะทางระบบประสาทมักสังเกตความอึดอัดใจของมอเตอร์และปฏิกิริยาทางระบบประสาท: ความกลัว, ความวิตกกังวล, กระสับกระส่าย, ฝันร้าย, การตอบสนองที่เพิ่มขึ้น, การชักที่มีลักษณะบาดทะยักในธรรมชาติและเกิดจากภาวะน้ำตาลในเลือดต่ำ, บางครั้งอาการชักกระตุก มีการอธิบายอาการของ Myopathic ด้วย: กล้ามเนื้ออ่อนแรง, กล้ามเนื้ออ่อนแรง มักพบความผิดปกติของ Extrapyramidal: choreiform hyperkinesis, athetosis, ใบหน้าซีก, พาร์กินสัน, ในบางกรณีมีโรคลมบ้าหมู paroxysms, อาการสมองน้อย: ataxia สูญเสียการประสานงาน
การกลายเป็นปูนของเนื้อเยื่ออ่อน มักตรวจพบการกลายเป็นปูนใต้ผิวหนัง (หน้าอก หน้าท้อง เอ็นส้นเท้า) ด้วย การตรวจชิ้นเนื้อที่ - โรคกระดูกพรุน(Izraeli et al., 1992), สมอง (ฐานปมประสาท) สิ่งสำคัญคือต้องสังเกตว่าอาจมีแคลเซียมเกิดขึ้นตั้งแต่แรกเกิด ผลจากภาวะแคลเซียมในเลือดต่ำ มักเกิดต้อกระจกและเคลือบฟันบกพร่อง
ซูโดไฮโปพาราโธซิสประเภท 1A
มีรูปแบบการถ่ายทอดลักษณะเด่นแบบออโตโซม ยีนสำหรับ pseudohypoparathyroidism ประเภท 1A - GNAS1 - มีการแปลเป็นภาษาท้องถิ่นบนแขนยาวของโครโมโซม 20 ในตำแหน่ง 20q13.2 การพัฒนาของโรคเกี่ยวข้องกับการขาดโปรตีนที่จับกับนิวคลีโอไทด์กัวนีน (โปรตีน Gs) ในเวลาเดียวกัน PTH ซึ่งจับกับตัวรับของเนื้อเยื่อเป้าหมาย ไม่สามารถกระตุ้นอะดีโนซีนโมโนฟอสเฟตแบบไซคลิก (cAMP) และทำให้เกิดการตอบสนองของเนื้อเยื่อได้ อาจเป็นกลไกที่คล้ายกันที่รองรับการพัฒนาของเนื้อเยื่อของอวัยวะอื่น ๆ และต่อมไร้ท่อ (hypofunction ของต่อมไทรอยด์, อวัยวะสืบพันธุ์, ต่อมใต้สมอง, โรคเบาหวานเช่นเดียวกับการตอบสนองที่ลดลงของตับต่อการบริหารกลูคากอน) พบใน pseudohypoparathyroidism ประเภท 1A ในพยาธิวิทยาประเภทนี้ ไม่พบการขับถ่าย cAMP ในปัสสาวะเพิ่มขึ้นตามปกติเพื่อตอบสนองต่อการบริหาร PTH จากภายนอก โรคนี้จะได้รับการวินิจฉัยบ่อยขึ้นเมื่ออายุ 5-10 ปี ผู้ป่วยมีรูปร่างเตี้ย คอสั้น ใบหน้ากลม กระดูกฝ่ามือและกระดูกฝ่าเท้าสั้นลง (โดยปกติจะสั้นลงของนิ้วที่สี่และน้อยกว่านิ้วที่สอง) - ที่เรียกว่า brachy-metaphalangism มีการสังเกตการกลายเป็นปูนของเนื้อเยื่ออ่อนและการกลายเป็นปูนใต้ผิวหนังซึ่งสามารถตรวจพบได้ตั้งแต่แรกเกิด มักพบการมีส่วนร่วมของต่อมไร้ท่ออื่น ๆ พร้อมกัน: ต่อมไทรอยด์ (hypofunction), อวัยวะสืบพันธุ์, ตับอ่อน (เบาหวาน) อันเป็นผลมาจากภาวะแคลเซียมในเลือดต่ำมักเกิดต้อกระจกและข้อบกพร่องของเคลือบฟัน เป็นการทดสอบการวินิจฉัยแยกโรคเพื่อแยกความแตกต่าง PHP ประเภท 1A จากภาวะพาราไทรอยด์ฮอร์โมนต่ำ: การไม่มีผลทางคลินิกจากการบริหาร PTH ทางหลอดเลือดดำในรูปแบบของการเพิ่มขึ้นของระดับแคลเซียมในเลือดและการขับถ่ายฟอสฟอรัสในไตเพิ่มขึ้นในปัสสาวะ (ผลฟอสฟอรัส)
การตรวจทางชีวเคมีเผยให้เห็นภาวะแคลเซียมในเลือดต่ำ, ภาวะฟอสเฟตในเลือดสูง, ระดับฮอร์โมนพาราไธรอยด์ในเลือดเพิ่มขึ้น และภาวะฟอสฟอรัสในเลือดต่ำ ระดับโปรตีน Gs ในเลือดลดลง ที่ การตรวจเอ็กซ์เรย์ระบบโครงร่างเผยให้เห็นการที่กระดูกฝ่ามือและกระดูกฝ่าเท้าสั้นลง การขาดแร่ธาตุโดยทั่วไป และกระดูกที่หนาขึ้นของกะโหลกโค้ง
โรคซูโดไฮโปพาราโธรซิสประเภท 1B
มีประเภทมรดกที่โดดเด่นแบบออโตโซม แต่ไม่มีการยกเว้นประเภทมรดกประเภท X-linked ที่โดดเด่น จำเป็นต้องคำนึงถึงการแทรกซึมของยีนของโรคที่ไม่สมบูรณ์ในบางครั้งและความเป็นไปได้ของการขนส่งทางพยาธิวิทยาที่แฝงอยู่ ดังนั้นจึงแนะนำให้ทำการตรวจทางคลินิก (การตรวจหาอาการไม่แสดงอาการ) และการตรวจทางชีวเคมี (การตรวจแคลเซียม ฟอสฟอรัส ระดับ PTH ในเลือด) ของผู้ให้บริการที่น่าสงสัย PHP ประเภท 1B เกิดจากการขาดตัวรับเนื้อเยื่อของฮอร์โมนพาราไธรอยด์ในอวัยวะเป้าหมาย และการดื้อต่อฮอร์โมนพาราไธรอยด์ที่จำกัด ภาพทางคลินิกคล้ายกับประเภท 1A แต่ไม่มีความเสียหายต่อต่อมไร้ท่ออื่นๆ และโรคกระดูกเสื่อมพบได้น้อย
ผู้ป่วยไม่มีการตอบสนองของไตต่อการบริหารฮอร์โมนพาราไธรอยด์จากภายนอกในรูปแบบของการขับถ่ายของอะดีโนซีนโมโนฟอสเฟตแบบไซคลิกเพิ่มขึ้นในปัสสาวะ อย่างไรก็ตาม ไม่เหมือนกับประเภท 1A ระดับของโปรตีน Gs ในเลือดเป็นปกติ ผู้หญิงได้รับผลกระทบบ่อยกว่าผู้ชาย แต่ความรุนแรงของโรคอาจแตกต่างกันทั้งในชายและหญิง
ซูโดไฮโปพาราโธรซิสประเภท 1C
ผู้เขียนบางคนระบุด้วย pseudo-pseudohypoparathyroidism (PPHP) ซึ่งบรรยายโดย Albright F. ในปี 1952 มีลักษณะเป็นภาพทางคลินิกของ PHP แต่ระดับแคลเซียมและฟอสฟอรัสในเลือดและปัสสาวะยังคงอยู่ในเกณฑ์ปกติ ระดับโปรตีน PTH และ Gs ในเลือดยังคงไม่เปลี่ยนแปลงเช่นกัน ระดับปกติ. ผู้ป่วยบางรายที่มี PHP ประเภท 1C มีการลบออก เดโนโวบนโครโมโซม 2 มีความเป็นไปได้ว่าแวเรียนท์ของโรคนี้เป็นชนิดย่อยของ PGP ชนิด 1A
โรคเทียมเทียมประเภท 2
ทางคลินิกคล้ายคลึงกับโรคชนิดอื่น แต่มีรูปแบบการถ่ายทอดทางพันธุกรรมแบบถอยอัตโนมัติ การมีอยู่ของรูปแบบพยาธิวิทยาที่โดดเด่นของออโตโซมไม่สามารถตัดออกได้ การเกิดโรคจากพัฒนาการสัมพันธ์กับการดื้อต่อแคมป์ภายในเซลล์ จากนั้น PTH จะจับกับตัวรับและทำให้เกิดการตอบสนองของเซลล์ตามปกติต่อ PTH ในรูปแบบของการขับถ่าย cAMP ที่เพิ่มขึ้น อย่างไรก็ตามความไม่รู้สึกภายในเซลล์ต่อแคมป์ไม่อนุญาตให้รับรู้ถึงผลกระทบเต็มรูปแบบของ PTH ในขณะเดียวกันก็ยังคงอยู่ ปฏิกิริยาปกติไตเพื่อการบริหารฮอร์โมนพาราไธรอยด์จากภายนอกในรูปแบบของการขับถ่ายที่เพิ่มขึ้นของอะดีโนซีนโมโนฟอสเฟตแบบไซคลิกในปัสสาวะ มีการแนะนำว่า PHP ประเภท 2 อาจเกี่ยวข้องกับการขาดวิตามินดี
ดังนั้นประเภท PGP ที่ระบุจึงมีลักษณะทางคลินิกโดยการลดความไวของอวัยวะเป้าหมายต่อ PTH แต่แตกต่างกันในกลไกการทำให้เกิดโรคของการก่อตัวของเนื้อเยื่อที่ไม่รู้สึก
การวินิจฉัย
การทดสอบการวินิจฉัยแยกโรคในห้องปฏิบัติการอาจเป็นรูปแบบของการขับถ่ายไตของค่ายเพื่อตอบสนองต่อการบริหาร PTH: การขับถ่ายของค่ายเพิ่มขึ้นในประเภท 2 และไม่มีอยู่ในประเภท 1 การวินิจฉัยได้รับการยืนยันโดยการตรวจพบระดับนิวคลีโอไทด์กัวนีนที่ลดลง การจับโปรตีน (Gs โปรตีน) ในเลือด (โดยเฉลี่ย 1.5–2 เท่า) เมื่อเปรียบเทียบกับบรรทัดฐาน ภาวะแคลเซียมในเลือดต่ำมักจะรวมกับภาวะฟอสเฟตในเลือดสูงและภาวะฟอสเฟตทูเรีย ระดับ PTH สูงขึ้น ในประเภท 1C ระดับ PTH เป็นปกติ ซึ่งทำให้เกิดชื่อ "pseudohypoparathyroidism" การตรวจเอ็กซ์เรย์ของระบบโครงร่างเผยให้เห็นการหดตัวของกระดูกฝ่ามือและกระดูกฝ่าเท้า ซึ่งมักทำให้เกิดการสูญเสียแร่ธาตุโดยทั่วไป (โรคกระดูกพรุน) และกระดูกของกะโหลกศีรษะหนาขึ้น รูปแบบ dermatoglyphic แสดงการกระจัดของ axial palmar triradius
เกณฑ์การวินิจฉัย:
- ขนาดสั้น;
- หน้ากลม;
- ความล่าช้าทางประสาท การพัฒนาจิต;
- ความผิดปกติของโครงกระดูก
- แคลเซียมในเลือดต่ำ
- ฮอร์โมนพาราไธรอยด์ในเลือดสูง
- ลดการขับถ่ายฟอสเฟตและแคมป์ในปัสสาวะ
การรักษาและการป้องกัน
การรักษาภาวะแคลเซียมในเลือดต่ำประกอบด้วยการสั่งจ่ายแคลเซียมเสริมในปริมาณที่เพียงพอเพื่อรักษาความเข้มข้นของแคลเซียมในเลือดให้เป็นปกติ การบำบัดด้วยวิตามินดีมีความสำคัญอย่างยิ่ง ปัจจุบันมีการใช้สารออกฤทธิ์ของวิตามินดี - ออกไซด์วิต, 1-อัลฟา-ดี3, แคลซิทริน ฯลฯ ในขนาด 1-2 ไมโครกรัมต่อวันด้วย ผลลัพธ์ที่เป็นบวก(เพิ่มระดับแคลเซียมในเลือด อาการลดลง อาการหงุดหงิด). ทาชิสทีน (0.5–1.5 มก./วัน) ก็ใช้ได้ผลเช่นกัน ยาตัวนี้เพิ่มการดูดซึมแคลเซียมในลำไส้จึงช่วยเพิ่มระดับแคลเซียมในเลือด การรักษาด้วยยากันชักใช้เป็นการรักษาเสริม บน การพัฒนาทางปัญญาการรักษาไม่มีผลที่เห็นได้ชัดเจน แต่เมื่ออาการของโรคหดเกร็งลดลงก็พบว่ามีการถดถอย อาการทางระบบประสาท(ความผิดปกติของ subcortical, choreiform hyperkinesis, athetosis ฯลฯ ) เพื่อหลีกเลี่ยงการเตรียมวิตามินดีเกินขนาดจำเป็นต้องตรวจสอบความเข้มข้นของแคลเซียมในเลือดทุก 3-7 วันในช่วง 2 สัปดาห์แรกของการรักษาและทุกเดือนในช่วง 2-3 เดือนข้างหน้า เมื่อความเข้มข้นของแคลเซียมในเลือดถึงระดับคงที่แล้ว ก็เพียงพอที่จะตรวจดูทุกๆ 2-3 เดือน การรับประทานอาหารที่ถูกจำกัดด้วยฟอสฟอรัสช่วยให้ความเข้มข้นของฟอสฟอรัสและแคลเซียมในเลือดเป็นปกติ และกำจัดอาการของภาวะพาราไทรอยด์ทำงานเกินขั้นทุติยภูมิ ในกรณีที่ต่อมไร้ท่ออื่นไม่เพียงพอจะทำการบำบัดทดแทนด้วยฮอร์โมนที่เหมาะสม
การรักษาด้วยฮอร์โมนพาราไธรอยด์ไม่ได้ผล เพื่อบรรเทาอาการชักจะมีการให้สารละลายแคลเซียมคลอไรด์หรือแคลเซียมกลูโคเนต 10% ทางหลอดเลือดดำ รับประทาน - สารละลายแคลเซียมคลอไรด์ 5-10%, 1 ช้อนโต๊ะ 3-4 ครั้งต่อวัน: แคลเซียมกลูโคเนต, แคลเซียมแลคเตต - มากถึง 10 กรัมต่อวัน
พยากรณ์ตลอดชีวิตจะพิจารณาจากความรุนแรงของกลุ่มอาการชัก
การป้องกันโรคนี้ขึ้นอยู่กับข้อมูลจากการให้คำปรึกษาทางพันธุกรรมทางการแพทย์
การให้คำปรึกษาทางการแพทย์และพันธุกรรม
เมื่อทำการให้คำปรึกษาทางพันธุกรรมทางการแพทย์ ควรดำเนินการตามประเภทการถ่ายทอดทางพันธุกรรมที่โดดเด่นแบบออโตโซม และมีความเสี่ยงสูง (50%) ที่จะเป็นโรคนี้ซ้ำในครอบครัวที่มีรูปแบบการถ่ายทอดทางพันธุกรรม เพื่อระบุลักษณะของประเภทของมรดกจำเป็นต้องทำการตรวจสอบผู้ปกครองอย่างละเอียดเนื่องจากกลุ่มอาการสามารถแสดงออกได้น้อยที่สุด อาการทางคลินิก. ปัจจุบันการวินิจฉัยโรคทางอณูพันธุศาสตร์ได้รับการพัฒนาและปรับปรุงโดยการพิมพ์การกลายพันธุ์ของยีน GNAS1 บนโครโมโซมคู่ที่ 20 วิธีการวินิจฉัยโรคก่อนคลอดโดยทั่วไปและประเภทของโรคอยู่ระหว่างการพัฒนา
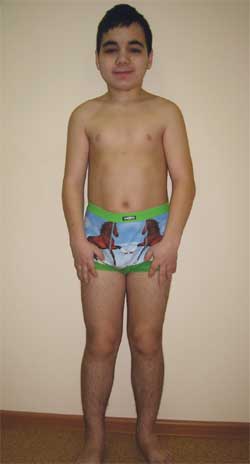
การสังเกตทางคลินิก Boy G. อายุ 14.5 ปี (รูปภาพ 4) เข้ารับการรักษาที่ Research Clinical Institute of Pediatrics ด้วยการวินิจฉัย: โรคความเสื่อม ระบบประสาท? hydrocephalus ภายนอกที่มีมา แต่กำเนิด; โรคลมบ้าหมูที่มีอาการ; โรคทางพันธุกรรม? โรคจากการสะสม? โรคสมองจากการเผาผลาญ; พร่องไม่แสดงอาการ; ความสูงสั้นของแหล่งกำเนิดผสม ความบกพร่องทางสติปัญญา
ร้องเรียนเมื่อเข้ารับการรักษาอาการปวดหัว paroxysmal ที่รุนแรงซึ่งแปลเป็นภาษาท้องถิ่นในบริเวณหน้าผากและมาพร้อมกับการอาเจียนซึ่งช่วยบรรเทาอาการความจำลดลงและประสิทธิภาพในโรงเรียนการโจมตีแบบชักในระหว่างที่มีการกระตุกเกิดขึ้นที่มือขวา
รูปภาพที่ 4
เด็ก G. อายุ 14.5 ปี มีภาวะกระดูกเสื่อม Albright
(ลักษณะฟีโนไทป์ รูปร่างเตี้ย แขนขาสั้น brachydactyly)
ประวัติครอบครัว:พ่อแม่เป็นชาวอาร์เมเนียตามสัญชาติ ไม่เกี่ยวข้องทางสายเลือด และไม่มีอันตรายทางวิชาชีพ ในสายเลือดของคดี ป่วยทางจิต, โรคลมบ้าหมู พัฒนาการล่าช้าไม่ได้สังเกต พี่สาวอายุ 17 ปี บอกว่าสุขภาพแข็งแรง
ประวัติความเป็นมาและความเจ็บป่วย:เด็กชายจากการตั้งครรภ์ครั้งที่ 2 ซึ่งดำเนินไปโดยไม่มีลักษณะเฉพาะใด ๆ การคลอดครั้งที่สองเมื่อครบกำหนดทางสรีรวิทยา น้ำหนักแรกเกิด - 3100 กรัม ความยาว - 51 ซม. เขาร้องออกมาทันที คะแนน Apgar - 7/9 คะแนน อาการแย่ลงวันที่ 3 – ทารกแรกเกิดมีอาการชัก พักรักษาตัวในโรงพยาบาลคลอดบุตร ช่วงหลังคลอดตอนต้นไม่มีคุณสมบัติ มีความล่าช้าเล็กน้อยในการพัฒนามอเตอร์ในปีแรกของชีวิตเดินอิสระตั้งแต่ 1 ปี 3 เดือน เกี่ยวกับเรื่องนี้ นักประสาทวิทยาได้รับการวินิจฉัยว่า: แผลอินทรีย์ระบบประสาทส่วนกลาง; hydrocephalus แต่กำเนิด; อาการชักของทารกแรกเกิด ประวัติอาการชักไข้
ฉันได้รับไดคาร์บและฟินเลปซิน เริ่มโจมตีเมื่ออายุ 1 ปี 11 เดือน – ไม่สมมาตร โทนิคในรูปแบบของความตึงเครียด มือขวาและขาโดยลืมตาได้นานถึง 2 นาทีโดยไม่หมดสติ บ่อยมากถึง 10 ตอนต่อวัน ฉันได้รับ Depakine อย่างไม่สม่ำเสมอ เมื่อเทียบกับพื้นหลังของการถอนตัวที่เป็นอิสระ - สถานะยาชูกำลังเดียว เมื่ออายุ 2 ปี มีการทำ CT scan ของสมอง ณ สถานที่อยู่อาศัย โดยมีการระบุจุดโฟกัสเดียวของการทำลายเยื่อในสมองกลีบท้ายทอย
ปรึกษาศัลยแพทย์ระบบประสาทและแนะนำให้รักษาแบบอนุรักษ์นิยม ตั้งแต่อายุ 3 ขวบ มีพัฒนาการทางจิตและการพูดล่าช้า แนะนำให้จิตแพทย์สังเกตอาการ
ตั้งแต่อายุ 4-5 ปี ผู้ปกครองเริ่มสังเกตเห็นการเสียรูปและการหดตัวของนิ้วและนิ้วเท้า โดยเฉพาะนิ้วที่ 2-4 ที่แขนและขาสมมาตรกัน และตัวชี้วัดการเจริญเติบโตลดลง เมื่ออายุ 8 ขวบ ข้อสรุปของนักบำบัดการพูดคือความผิดปกติในการพูดทั่วไปในระดับ 2-3 แนะนำให้ศึกษาในโรงเรียนเฉพาะทาง ในวัยเดียวกัน ตรวจโดยนักพันธุศาสตร์ ณ สถานที่อยู่อาศัย สรุปว่า โรคทางพันธุกรรมแลกเปลี่ยน? แนะนำให้ศึกษากรดอะมิโนในเลือด ไม่พบการเปลี่ยนแปลง ข้อสรุปสุดท้าย: ไม่พบหลักฐานของโรคเมตาบอลิซึมทางพันธุกรรม ภาวะ hypochondroplasia; แนะนำให้ทำการรักษาโดยนักประสาทวิทยาและแพทย์ต่อมไร้ท่อ
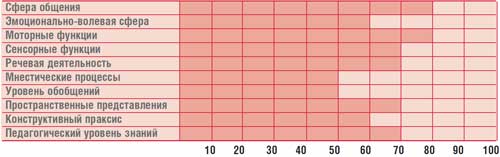
โต๊ะ.
ประวัติพัฒนาการทางจิตของเด็ก ก. อายุ 14.5 ปี (IQ = 68)
เมื่ออายุ 8 ปี เขาได้รับคำปรึกษาจากแพทย์ด้านต่อมไร้ท่อเกี่ยวกับการเจริญเติบโตและการพัฒนาที่ล่าช้า การตรวจเอ็กซ์เรย์มือเผยให้เห็นลักษณะต่อไปนี้: กระดูกส่วนตรงกลางและส่วนหลักและกระดูกฝ่ามือนั้นสั้นและหนาขึ้น การวินิจฉัยของนักรังสีวิทยาคือ achondroplasia
ตรวจซ้ำ ณ สถานที่อยู่อาศัยในโรงพยาบาลระบบประสาท เมื่ออายุ 12 ปีการโจมตีแบบชักปรากฏขึ้นโดยไม่หมดสติด้วยการกระตุกที่แขนขวาซึ่งมีลักษณะต่อเนื่องกัน มีการกำหนดการรักษาด้วยยากันชัก (Depakine) และความถี่ของการโจมตีลดลงอย่างมีนัยสำคัญ เมื่ออายุ 13 ปี MRI ของสมองที่มีความคมชัดได้ดำเนินการ - การเปลี่ยนแปลงแบบสมมาตรในฐานของกลีบขมับที่ระดับนิวเคลียสในรูปแบบของการเพิ่มขึ้นของสัญญาณ MR ซึ่งเป็นเรื่องปกติสำหรับพิษ (แมงกานีส) หรือโรคสมองจากการเผาผลาญ (ทองแดง, เหล็ก)
ตรวจอีกครั้งเมื่ออายุ 13 ปี 3 เดือน ต่อมไร้ท่อ การศึกษาโปรไฟล์ของต่อมไทรอยด์พบว่ามีการเพิ่มขึ้น ฮอร์โมนกระตุ้นต่อมไทรอยด์(TSH), วินิจฉัยภาวะพร่องไทรอยด์แบบไม่แสดงอาการ, กำหนดให้ L-thyroxine
เมื่อวิเคราะห์บัตรผู้ป่วยนอกและเอกสารประกอบของเด็ก ณ สถานที่อยู่อาศัย การศึกษาแคลเซียมและฟอสฟอรัสได้ดำเนินการหนึ่งครั้งเมื่ออายุ 1.5 ปี พบว่ามีภาวะน้ำตาลในเลือดต่ำ แต่ไม่มีการตรวจเพิ่มเติมในครั้งนี้ เมื่อพิจารณาถึงความไม่แน่นอนของการวินิจฉัย ณ สถานที่อยู่อาศัย นักพันธุศาสตร์จึงส่งเด็กไปมอสโคว์ไปที่สถาบันวิจัยคลินิกกุมารเวชศาสตร์ทางวิทยาศาสตร์เพื่อชี้แจงการวินิจฉัย
ข้อมูล การวิจัยตามวัตถุประสงค์:
ส่วนสูง – 143 ซม. น้ำหนัก – 43 กก.
พัฒนาการทางร่างกายต่ำมาก ความสามัคคี ร่างกายไม่สมส่วนเนื่องจากแขนขาสั้นลง Growth Sds สอดคล้องกับ –2.8 ส่วนเบี่ยงเบนจากบรรทัดฐาน (บรรทัดฐาน –2+2)
ลักษณะฟีโนไทป์: หน้ากลม, คอสั้น, รอยแยกของเปลือกตามองโกลอยด์, สะพานจมูกกว้าง, หน้าผากสูง, brachydactyly, การทำให้กระดูกฝ่าเท้า IV และ V และกระดูกฝ่าเท้าสั้นลง (รูปภาพ 5) อวัยวะภายใน – ไม่มีลักษณะเฉพาะใดๆ พัฒนาการทางเพศ – แทนเนอร์ระยะที่ III–IV (ซึ่งสอดคล้องกับอายุ)
ห้องปฏิบัติการและ การศึกษาเชิงหน้าที่:
การวิเคราะห์ทางคลินิกเลือดและปัสสาวะเป็นเรื่องปกติ
การวิเคราะห์ทางชีวเคมีเลือด: แคลเซียมทั้งหมด– 1.39 (ปกติ 2.02–2.6 มิลลิโมล/ลิตร) แคลเซียมแตกตัวเป็นไอออน – 0.61 (ปกติ 1.13–1.32 มิลลิโมล/ลิตร) ฟอสฟอรัสอนินทรีย์ – 3.66 (ปกติ 0.86–1.56 มิลลิโมล/ลิตร) ตัวชี้วัดอื่นๆ อยู่ภายในขีดจำกัดปกติ
การวิเคราะห์ปัสสาวะทางชีวเคมี: การขับถ่ายฟอสเฟตในไตลดลง - 11.5 มิลลิโมล/ลิตร (ปกติ 19–32 มิลลิโมล/ลิตร)
โปรไฟล์ของต่อมไทรอยด์: TSH – 11.75 (ช่วงปกติ 0.4–4.0 µIU/ml), T4 ฟรี – 0.49 (ช่วงปกติ 1.0–1.8 ng/dL)
ฮอร์โมนพาราไธรอยด์ – 499 (ปกติ 12–65 pg/ml), STH – 7 ng/ml (ปกติ 7–10 ng/ml), somatomedin-C – 250 ng/ml (ปกติ 88–360 ng/ml)
อัลตราซาวนด์ อวัยวะภายใน- ไม่มีคุณสมบัติ
ECG - การโยกย้ายของเครื่องกระตุ้นหัวใจเหนือหัวใจโดยมีอัตราการเต้นของหัวใจปกติอยู่ที่ 71–80 ครั้งต่อนาที การปิดล้อมสาขามัดด้านขวาไม่สมบูรณ์ การรบกวนกระบวนการรีโพลาไรซ์ในกล้ามเนื้อหัวใจ ผนังด้านหลังช่องซ้าย (ลดลง zT III, aVF)
R-graphy ของกระดูกสันหลัง – scoliosis ทางด้านขวา ทรวงอกกระดูกสันหลังระดับ 1 โรคกระดูกพรุนรุนแรง
ภาพ R ของมือพร้อมการจับปลายแขน - การย่อและขยายของส่วนปลายและช่วงกลาง อายุกระดูก – 13.5–14 ปี
ไม่มีการบันทึกรูปแบบ EEG ของการเกิดโรคลมบ้าหมู
MRI ของสมอง - ภาพ MR ของจุดโฟกัส subcortical หลายจุดของสัญญาณ MR ที่เพิ่มขึ้นในกลีบหน้าผาก, hydrocephalus ชดเชยภายนอกที่มีการฝ่อของสารในสมอง
MSCT ของสมองแสดงพื้นที่ที่สมมาตรของการกลายเป็นปูนของนิวเคลียสของถั่วเลนติฟอร์ม กระจายพื้นที่ที่มีความหนาแน่นสูงในทาลามิ นิวเคลียสหางที่มีพื้นที่กลายเป็นปูนทางด้านขวา การกลายเป็นปูนหลายจุดของเนื้อเยื่ออ่อนของกะโหลกศีรษะ
Audiogram – ไม่มีพยาธิวิทยา
การวินิจฉัย DNA ในยีน GNAS1 อยู่ระหว่างดำเนินการ
การให้คำปรึกษาผู้เชี่ยวชาญ:
แพทย์ต่อมไร้ท่อ – โรคกระดูกพรุนทางพันธุกรรมของอัลไบรท์ประเภท 1A (pseudohypoparathyroidism) พร่องปฐมภูมิ,ค่าชดเชยยาไม่ครบถ้วน
จักษุแพทย์ – ต้อกระจกทุติยภูมิที่สมบูรณ์ ที่แนะนำ การผ่าตัดรักษา.
นักจิตวิทยา – ความบกพร่องทางสติปัญญา (รายละเอียดทางจิตวิทยาของเด็กแสดงอยู่ในตาราง)
โดยคำนึงถึงฟีโนไทป์ ประวัติการรักษา และผลลัพธ์ของเด็ก การวิจัยเพิ่มเติม(ภาวะแคลเซียมในเลือดต่ำ, ภาวะฟอสเฟตในเลือดสูง, ภาวะฟอสเฟตทูเรีย, ฮอร์โมนพาราไธรอยด์ในเลือดเพิ่มขึ้น), การกลายเป็นปูนในสมอง, การปรากฏตัวของต้อกระจก, ภาวะไทรอยด์ทำงานต่ำ), มีการวินิจฉัย: โรคกระดูกพรุนทางพันธุกรรมของออลไบรท์ประเภท 1A (pseudohypoparathyroidism) ขอแนะนำให้ทำการวินิจฉัย DNA - ค้นหาการกลายพันธุ์ในยีน GNAS1
การรักษา:แนะนำให้เด็กรับประทาน eutirox ในขนาด 100 ไมโครกรัมต่อวัน สารออกฤทธิ์ของวิตามินดี - alpha-D3 (Teva) ในขนาด 2 ไมโครกรัมต่อวัน; แคลเซียม (แซนดอซ) 2,000 มก./วัน; การใช้การรักษาด้วยยากันชักอย่างต่อเนื่อง - finlepsin 800 มก. / วันภายใต้การดูแลของนักประสาทวิทยา - โรคลมชัก; ชั้นเรียนกับนักพยาธิวิทยาด้านการพูดและนักจิตวิทยา การบำบัดด้วยพลังงานเขตร้อน (Elkar และโคเอ็นไซม์คิว 10 ในปริมาณที่เกี่ยวข้องกับอายุ) การติดตามตัวชี้วัดการเผาผลาญฟอสฟอรัส-แคลเซียม ระดับฮอร์โมนพาราไธรอยด์
ดังนั้น,การสังเกตทางคลินิกที่นำเสนอแสดงให้เห็นถึงความยากลำบากในการค้นหาการวินิจฉัยแยกโรค ความสำคัญของการศึกษาพารามิเตอร์ทางชีวเคมีอย่างง่ายอย่างทันท่วงที (ในกรณีของโรคลมบ้าหมู จำเป็นต้องมีการตรวจคัดกรองตัวชี้วัดการเผาผลาญฟอสฟอรัส-แคลเซียมซ้ำ) ผลลัพธ์ของการวินิจฉัยโรคที่กำหนดทางพันธุกรรมล่าช้า ความจำเป็นในการบูรณาการสัญญาณส่วนบุคคลเข้ากับฟีโนไทป์โดยรวมของโรคเฉพาะ สภาพทางพยาธิวิทยาเพื่อการวินิจฉัยแต่ละรูปแบบอย่างตรงเป้าหมายและทันท่วงที โรคทางพันธุกรรม. การวินิจฉัยและการชี้แจงการกำเนิดของแต่ละกลุ่มอาการอย่างทันท่วงทีมีความสำคัญอย่างยิ่งเนื่องจากช่วยให้เราสามารถค้นหาแนวทางที่เหมาะสมที่สุดในการรักษาเงื่อนไขและการป้องกันเหล่านี้ ภาวะแทรกซ้อนที่เป็นไปได้(ขึ้นอยู่กับความพิการของเด็ก) การป้องกันการกลับเป็นซ้ำของโรคทางพันธุกรรมในครอบครัวที่ได้รับผลกระทบ (การให้คำปรึกษาทางการแพทย์และทางพันธุกรรม) สิ่งนี้กำหนดความจำเป็นสำหรับแพทย์ที่เชี่ยวชาญด้านต่างๆ เพื่อนำทางการไหลของพยาธิวิทยาที่กำหนดโดยกรรมพันธุ์อย่างชัดเจน รายการข้อมูลอ้างอิงอยู่ในกองบรรณาธิการ