
biokimia fungsional. Porfiria - penyebab, klasifikasi biokimia Porfiria
Jenis-Jenis Porfiria - Kuliah, Bagian Kimia, KURSUS KURSUS BIOKIMIA UMUM Porfiria Intermiten Akut (Opp) - Penyebab - Cacat Ge.
Porfiria intermiten akut (API) - penyebabnya adalah cacat pada gen penyandi PBG - deaminase.
Diwariskan secara autosomal dominan. Ada akumulasi prekursor awal sintesis heme: 5-ALA (5-ALA) dan porfobilinogen (PBG).
Porfiria eritropoietik kongenital - jarang terjadi penyakit bawaan diturunkan secara autosomal resesif. Hal ini ditandai dengan aktivitas uroporfirinogen-III-kosintase yang rendah dan aktivitas uroporfirinogen-I-sintase yang tinggi. Pembentukan uroporfirinogen-I secara signifikan melebihi sintesis uroporfirinogen-III (isomer normal dalam jalur sintesis heme). Meskipun kelainan genetik meluas ke semua sel, itu memanifestasikan dirinya, untuk alasan yang tidak diketahui, terutama di jaringan eritropoietik. Pasien mengeluarkan sejumlah besar uroporfirinogen-I dan koproporfirinogen-I dalam urin; yang secara spontan teroksidasi di udara menjadi uroporfirin-I dan koproporfirin-I - pigmen fluoresen merah. Eritrosit yang bersirkulasi mengandung sejumlah besar uroporfirin-I, namun konsentrasi tertinggi porfirin ini tercatat dalam sel. sumsum tulang.
Ada sensitivitas cahaya pada kulit karena aktivasi peroksidasi lipid di bawah aksi cahaya pada senyawa porfirin. Pasien memiliki retakan pada kulit, dan fenomena hemolitik sering diamati.
Koproporfiria herediter adalah kelainan dominan autosomal yang disebabkan oleh defisiensi koproporfirinogen oksidase, suatu enzim mitokondria yang bertanggung jawab atas konversi koproporfirinogen III menjadi protoporfirinogen IX. Koproporfirinogen III diekskresikan dalam jumlah besar dari tubuh melalui feses dan urin. Koproporfirinogen dalam cahaya dan udara dengan cepat teroksidasi, berubah menjadi pigmen merah koproporfirin.
Kemampuan terbatas untuk mensintesis heme (terutama dalam kondisi stres) menyebabkan derepresi ALA sintase. Akibatnya, pembentukan berlebihan ALA, porfobilinogen dan zat antara lain dari sintesis heme diamati. Pasien menunjukkan semua tanda dan gejala yang berhubungan dengan kelebihan ALA dan porfobilinogen yang merupakan karakteristik porfiria akut intermiten, ada peningkatan fotosensitifitas karena adanya jumlah koproporfirinogen dan uroporfirinogen yang berlebihan.
Porfiria mosaik, atau fotoporfiria herediter, adalah kelainan dominan autosomal di mana ada penyumbatan parsial konversi enzimatik protoporfirinogen menjadi heme. Biasanya, transformasi ini dilakukan oleh dua enzim, protoporfirinogen oksidase dan ferrochelatase, yang terlokalisasi di mitokondria. Kandungan protoporfirinogen oksidase hanya setengah dari jumlah normal. Pada pasien dengan porfiria mosaik, kandungan heme relatif berkurang dalam kondisi stres, serta keadaan ALA sintase hepatik yang tertekan. Hal ini menyebabkan kelebihan produksi semua zat antara sintesis heme di area sebelum tahap yang diblokir. Pasien dengan porfiria mosaik mengekskresikan ALA, porfobilinogen, uroporfirin, dan koproporfirin dalam jumlah berlebih dalam urin, dan mengekskresikan uroporfirin, koproporfirin, dan protoporfirin dalam tinja. Urin pasien berpigmen dan berfluoresensi, dan kulit sensitif terhadap cahaya dengan cara yang sama seperti pada pasien dengan porfiria kutaneous tardive (lihat di bawah).
Porfiria tardio kulit adalah bentuk porfiria yang paling umum. Hal ini biasanya berhubungan dengan kerusakan hati, terutama dengan asupan alkohol yang berlebihan atau kelebihan ion besi. Kemungkinan penyebabnya adalah defisiensi parsial uroporfirinogen dekarboksilase. Gangguan tersebut tampaknya ditransmisikan sebagai autosomal sifat dominan, tetapi penetrasi genetik bervariasi dan dalam banyak kasus tergantung pada adanya fungsi hati yang abnormal. Urine mengandung uroporfirin tipe I dan III dalam jumlah tinggi; pada saat yang sama, ekskresi urin ALA dan porfobilinogen relatif jarang. Terkadang urin mengandung porfirin dalam jumlah yang sangat signifikan, memberikan warna merah muda; ketika diasamkan, paling sering memberikan fluoresensi merah muda di wilayah ultraviolet.
Topik ini milik:
KURSUS KULIAH TENTANG BIOKIMIA UMUM
Departemen Biokimia. KURSUS KULIAH TENTANG BIOKIMIA UMUM.
Jika Anda membutuhkan material tambahan pada topik ini, atau Anda tidak menemukan apa yang Anda cari, kami sarankan menggunakan pencarian di database karya kami: Jenis porfiria
Apa yang akan kami lakukan dengan materi yang diterima:
Jika materi ini ternyata bermanfaat bagi Anda, Anda dapat menyimpannya ke halaman Anda di jejaring sosial:
Semua topik di bagian ini:
Darah adalah jaringan cair yang melakukan sejumlah fungsi penting dalam tubuh: Fungsi utama darah adalah pengangkutan zat dan energi panas; a.Fungsi pernafasan
Semua cairan tubuh memiliki sifat umum: volume, densitas, viskositas, pH dan tekanan osmotik. Nilai normal sifat umum darah orang dewasa:
Elemen yang terbentuk (sel) darah membentuk 45% dari total volume darah. No Sel darah Konsentrasi % dari total volume ke
Komposisi kimia zat yang larut dalam plasma darah relatif konstan, karena ada mekanisme saraf dan humoral yang kuat yang mempertahankan homeostasis.
Indikator Usia 1 hari 1 bulan 6 bulan 1 tahun 1-6 L 12 L
Lebih dari 200 jenis protein telah ditemukan dalam plasma darah, yang membentuk 7% dari volume plasma. Protein plasma disintesis terutama di hati dan makrofag, serta di endotel vaskular, di usus, getah bening.
Secara struktur, protein plasma darah berbentuk globular; berdasarkan komposisi, mereka dibagi menjadi sederhana (albumin) dan kompleks. Di antara yang kompleks, lipoprotein (VLDL, LPP, LDL, HDL, HM), gli
Protein utama dari fraksi ini adalah albumin. albumen. Protein sederhana 585 AA dengan massa 69 kDa, memiliki 17 jembatan disulfida, banyak AA dikarboksilat, memiliki tinggi
VLDL - terbentuk di hati. Transportasi TG, XC. LPPP - terbentuk dalam darah dari VLDL. Transportasi TG, XC.
Disintesis oleh limfosit B yang aktif secara fungsional (plasmosit). Orang dewasa memiliki 107 klon limfosit B yang mensintesis 107 jenis -globulin. -bola
Konsep "protein fase akut" menggabungkan hingga 30 protein plasma darah yang terlibat dalam respons inflamasi tubuh terhadap cedera. Protein fase akut disintesis di hati, ujungnya
Enzim yang terdapat dalam plasma darah dapat dibagi menjadi 3 kelompok utama: 1. Sekretori. Mereka disintesis di hati, endotel usus, pembuluh darah masuk
Fakultas: medis dan preventif, medis dan preventif, pediatrik. 2 kursus. Eritrosit (eritrositus) adalah elemen berbentuk darah,
Eritrosit pada manusia dan mamalia dalam aliran darah biasanya (80%) berbentuk cakram bikonkaf dan disebut diskosit. Bentuk eritrosit ini menciptakan area terbesar
Oligosakarida (asam sialic dan oligosakarida antigenik) dari glikolipid dan glikoprotein yang terletak di permukaan luar membran plasma membentuk glikokaliks.
Dalam eritrosit matang, protein tidak disintesis, karena tidak memiliki ribosom, EPR, aparatus Golgi, dan nukleus. Namun, glutathione peptida disintesis di sitoplasma. Biosintesis glutathione dilakukan di
Dalam eritrosit dewasa: 1. AMP dapat disintesis dari FRPP (dari ribosa-5f) dan adenin. 2. AMP dengan partisipasi ATP diubah menjadi ADP. 3. Dalam reaksi fosforilasi substrat
Pada siang hari, hingga 3% hemoglobin dapat dioksidasi secara spontan menjadi methemoglobin: Hb (Fe2+) ® Met Hb (Fe3+) + e- Pemulihan methemoglobin menjadi hemoglobin
Heme adalah porfirin, di tengahnya adalah
Heme adalah kelompok prostat dari banyak protein: hemoglobin, mioglobin, sitokrom CPE mitokondria, sitokrom P450, enzim katalase, peroksidase, sitokrom oksidase, triptofan
Heme disintesis di semua jaringan, tetapi pada tingkat tertinggi di sumsum tulang dan hati. Di sumsum tulang, heme diperlukan untuk sintesis hemoglobin, di hepatosit - untuk pembentukan sitokrom P450
Heme yang disintesis di mitokondria menginduksi sintesis rantai globin pada poliribosom. Gen rantai globin terletak pada kromosom 11 dan 16. Rantai globin membentuk globul dan berikatan dengan he
Hemoglobin adalah kromoprotein tetramerik, memiliki massa 64,5 kDa, terdiri dari 4 heme dan 4 globin. Globin diwakili oleh rantai polipeptida dari berbagai jenis a, b, g, d, dll. sebuah rantai
Hemoglobin dengan ikatan besi koordinasi keenam bebas di heme disebut apohemoglobin. Ikatan koordinasi keenam dapat mengikat berbagai ligan
Hemoglobin mengikat O2 secara berurutan, satu molekul untuk setiap heme. Dalam apohemoglobin, karena ikatan koordinasi dengan bagian protein, atom
Kerja sama dalam kerja protomer hemoglobin membentuk karakter sigmoid dari kurva saturasi oksigen tergantung pada tekanan parsial oksigen. Kurva saturasi berbentuk S
Pengaruh pH pada sifat kurva disosiasi oksihemoglobin disebut efek Bohr (setelah ahli fisiologi Denmark Christian Bohr, yang pertama kali menemukan efek ini). &nb
2,3-DPG mengurangi afinitas hemoglobin untuk oksigen dan dengan demikian meningkatkan kembalinya oksigen ke jaringan. Jika darah telah menggunakan seluruh suplai DPG, hemoglobin tetap jenuh secara efektif dengan oksigen.
1) hemoglobin embrionik (Pengawas I, Pengatur II). Pada tahap awal perkembangan janin pada minggu-minggu pertama perkembangan, ketika fokus hematopoiesis muncul di kantung kuning telur,
Hemoglobin abnormal dihasilkan dari mutasi pada gen yang mengkode rantai a dan b. Beberapa ratus hemoglobin manusia mutan diketahui (aktif secara fungsional dalam banyak kasus).
Penyakit hemoglobin disebut hemoglobinosa, ada lebih dari 200 di antaranya.Hemoglobinosis dibagi menjadi hemoglobinopati dan talasemia. Hemoglobinopati
Eritrosit tua yang rusak difagositosis oleh sel RES dan dicerna dalam lisosom. Pemecahan hemoglobin menghasilkan pigmen empedu bilirubin. Katabolisme lebih lanjut dari bilirubin di hati, kish
Ingin menerima berita terbaru melalui email?
Berlangganan newsletter kami
Berita dan info untuk siswa
Periklanan
Materi terkait
- Serupa
- Populer
- Tandai Awan
- Di Sini
- Untuk sementara
- kosong
Tentang situs
Informasi dalam bentuk abstrak, abstrak, kuliah, makalah dan tesis memiliki penulisnya sendiri, yang memiliki hak. Oleh karena itu, sebelum menggunakan informasi apa pun dari situs ini, pastikan Anda tidak melanggar hak siapa pun dengan melakukannya.
Biokimia Porfiria
Heme, zat warna tetrahidropirol yang mengandung besi, merupakan komponen protein pengikat O2 (lihat hal. 274) dan berbagai koenzim oksidoreduktase (lihat hal. 108, 310). Hampir 85% biosintesis heme terjadi di sumsum tulang dan hanya sebagian kecil - di hati. Mitokondria dan sitoplasma terlibat dalam sintesis heme.
Sintesis cincin tetrahidropirol dimulai di mitokondria. Succinyl-CoA (diagram atas), zat antara dalam siklus sitrat, mengembun dengan glisin untuk menghasilkan produk yang didekarboksilat menjadi 5-aminolevulinate (ALA). Sintase 5-aminolevulinat (ALA sintase) yang bertanggung jawab untuk langkah ini [1] adalah enzim kunci dari keseluruhan jalur. Ekspresi sintesis ALA sintase dihambat oleh heme, yaitu produk akhir, dan enzim yang ada. Ini adalah kasus khas penghambatan oleh produk akhir, atau penghambatan umpan balik.
Setelah sintesis, 5-aminolevulinate berpindah dari mitokondria ke sitoplasma, di mana dua molekul memadat menjadi porfobilinogen, yang sudah mengandung cincin pirol [2]. Porfobilinogen sintase [2] dihambat oleh ion timbal. Oleh karena itu, ketika keracunan akut ditemukan timbal dalam darah dan urin konsentrasi tinggi 5-aminolevulinat.
Pada tahap selanjutnya, struktur tetrapirol karakteristik porfirin terbentuk. Pengikatan empat molekul porfobilinogen dengan eliminasi gugus NH2 dan pembentukan uroporfirinogen III dikatalisis oleh hidroksimetilbilane sintase [3]. Pembentukan produk antara ini membutuhkan enzim kedua, uroporfirinogen III sintase [4]. Tidak adanya enzim ini mengarah pada pembentukan isomer "salah" - uroporfirinogen I.
Struktur tetrapirol uroporfirinogen III masih berbeda secara signifikan dari heme. Jadi, tidak ada atom besi pusat, dan cincin itu hanya mengandung 8, bukan 11 ikatan rangkap. Selain itu, cincin hanya membawa rantai samping R bermuatan (4 residu asetat dan 4 propionat). Karena gugus heme dalam protein berfungsi dalam lingkungan non-polar, rantai samping polar perlu menjadi kurang polar. Pertama, empat residu asetat (R 1) didekarboksilasi untuk membentuk gugus metil (5). Koproporfirinogen III yang dihasilkan kembali ke mitokondria lagi. Langkah selanjutnya dikatalisis oleh enzim yang terlokalisasi pada/atau di dalam membran mitokondria. Pertama-tama, di bawah aksi oksidase, dua gugus propionat (R 2) diubah menjadi vinil (6). Modifikasi rantai samping berakhir dengan pembentukan protoporfirinogen IX.
Pada tahap berikutnya, karena oksidasi, sistem -elektron terkonjugasi dibuat dalam molekul, yang memberikan heme warna merah yang khas. Ini menghabiskan 6 ekuivalen pemulihan (7). Akhirnya, dengan bantuan enzim khusus, ferrochelatase, atom besi dimasukkan ke dalam molekul (8). Heme atau Fe-protoporfirin IX yang terbentuk dengan cara ini termasuk, misalnya, dalam hemoglobin dan mioglobin (lihat hal. 274), di mana ia terikat secara non-kovalen, atau dalam sitokrom C, yang mengikatnya secara kovalen (lihat hal. 108).
Ada sejumlah penyakit yang disebabkan oleh gangguan sintesis porfirin herediter atau didapat, yang disebut porfiria; beberapa di antaranya sangat sulit. Banyak dari penyakit ini mengakibatkan ekskresi prekursor heme dalam feses atau urin, yang karenanya dapat berwarna merah tua. Deposisi porfirin di kulit juga diamati. Saat terkena cahaya, ini mengarah pada pembentukan lepuh keras. Seringkali dengan porfiria kelainan saraf. Ada kemungkinan bahwa legenda abad pertengahan tentang orang vampir (drakula) didasarkan pada perilaku aneh pasien dengan porfiria (fotofobia, penampilan dan perilaku yang tidak biasa, makan darah, yang mengkompensasi kekurangan heme dan sering memperbaiki kondisi dalam beberapa bentuk. porfiria).
Hemoglobin memiliki penyakit molekuler
anemia sel sabit
HbS adalah hemoglobin sel sabit. Pada kelainan DNA ini, sebagai akibat dari mutasi titik, triplet CTT digantikan oleh triplet CAT, yang memerlukan masuknya asam amino valin alih-alih glutamat pada posisi ke-6 dari rantai . Perubahan sifat rantai menyebabkan perubahan sifat seluruh molekul dan pembentukan situs "lengket" pada permukaan hemoglobin. Selama deoksigenasi hemoglobin, situs "membuka" dan mengikat satu molekul deoksihemoglobin S ke molekul serupa lainnya. Hasilnya adalah polimerisasi molekul hemoglobin dan pembentukan untaian protein besar, menyebabkan deformasi eritrosit dan hemolisis selama perjalanan kapiler.
Skema perbedaan antara hemoglobin S dan hemoglobin A dan polimerisasinya
Pelanggaran sintesis hemoglobin
Porfiria
Porfiria adalah kelompok heterogen penyakit keturunan akibat pelanggaran sintesis heme dan peningkatan kandungan porfirin dan prekursornya dalam tubuh. Alokasikan bentuk porfiria herediter dan didapat.
Bentuk porfiria yang didapat bersifat toksik dan disebabkan oleh aksi heksaklorobenzena, garam timbal dan logam berat lainnya (penghambatan porfobilinogen sintase, ferrochelatase, dll.), obat-obatan (antibiotik antijamur griseofulfin).
Dalam bentuk herediter, cacat enzim hadir di semua sel tubuh, tetapi memanifestasikan dirinya hanya dalam satu jenis sel. Dua dapat dibedakan kelompok besar porfiria:
1. Hepatik - sekelompok penyakit dengan gangguan enzim autosomal dominan pada berbagai tahap sintesis protoporfirin IX. Penyakit yang paling mencolok dari kelompok ini adalah porfiria akut intermiten, di mana aktivitas uroporfirinogen-I-sintase pada heterozigot berkurang 50%.
Penyakit ini memanifestasikan dirinya setelah mencapai pubertas karena peningkatan kebutuhan hepatosit dalam sitokrom P 450 untuk menetralkan steroid seks, eksaserbasi kondisi juga sering terjadi setelah minum obat yang metabolismenya memerlukan partisipasi sitokrom P 450. Penurunan konsentrasi heme yang digunakan untuk sintesis sitokrom P 450 mengaktifkan aminolevulinate sintase. Akibatnya, pasien mengeluarkan sejumlah besar porfobilinogen dan asam aminolevulinat dalam urin. Dalam cahaya, porfirinogen dioksidasi menjadi porfobilin dan porfirin berwarna, dan inilah alasan urin menjadi gelap saat terkena cahaya dengan akses ke udara. Gejalanya adalah nyeri perut akut, konstipasi, gangguan kardiovaskular, gangguan neuropsikiatri.
2. Eritropoietik - gangguan resesif autosomal dari beberapa enzim sintesis protoporfirin IX dalam sel eritroid. Pada saat yang sama, keseimbangan reaksi untuk pembentukan uroporfirinogen bergeser ke arah sintesis uroporfirinogen I. Gejala penyakitnya mirip dengan yang sebelumnya, tetapi selain itu, fotosensitifitas kulit diamati karena adanya uroporfirinogen. , retakan pada kulit dan fenomena hemolitik dicatat.
Thalasemia
Thalassemia ditandai dengan penurunan sintesis hemoglobin rantai (α-thalassemia) atau rantai (β-thalassemia). Hal ini menyebabkan gangguan eritropoiesis, hemolisis dan anemia berat.
Anda dapat bertanya atau meninggalkan pendapat Anda.
Jenis-jenis porfiria
Gangguan sintesis heme. Porfiria
Porfiria adalah sekelompok penyakit heterogen yang disebabkan oleh gangguan sintesis heme karena kekurangan satu atau lebih enzim.
Tidak ada klasifikasi tunggal porfiria. Porfiria dibagi karena alasan menjadi:
1) turun temurun. Terjadi bila ada defek pada gen enzim yang terlibat dalam sintesis heme;
2) Diperoleh. Terjadi dengan efek penghambatan senyawa beracun (hexochlorobenzene, garam logam berat - timbal) pada enzim sintesis heme.
Tergantung pada lokalisasi dominan defisiensi enzim (di hati atau eritrosit), porfirin dibagi menjadi:
1) hati- jenis porfirin yang paling umum adalah porfiria intermiten akut (AKI), porfiria tardive kulit, koproporfiria herediter, porfiria mosaik;
2) eritropoietik- porfiria eritropoietik kongenital (penyakit Gunther), protoporfiria eritropoietik.
Tergantung pada Gambaran klinis, porfiria dibagi menjadi:
Konsekuensi negatif dari porfiria dikaitkan dengan defisiensi heme dan akumulasi dalam jaringan dan darah produk antara sintesis heme - porfirinogen dan produk oksidasinya. Pada porfiria eritropoietik, porfirin terakumulasi dalam normoblas dan eritrosit; pada porfiria hepatik, dalam hepatosit.
Untuk setiap jenis porfiria, ada tingkat cacat enzimatik tertentu, akibatnya, produk yang disintesis di atas tingkat ini menumpuk. Produk-produk ini adalah penanda diagnostik utama penyakit ini.
Porfirinogen beracun bentuk parah porfiria, mereka menyebabkan gangguan neuropsikiatri, disfungsi RES dan kerusakan kulit.
Gangguan neuropsikiatri pada porfiria dikaitkan dengan fakta bahwa aminolevulinat dan porfirinogen adalah neurotoksin.
Di kulit di bawah sinar matahari, porfirinogen mudah diubah menjadi porfirin. Oksigen, ketika berinteraksi dengan porfirin, masuk ke keadaan singlet. Oksigen singlet merangsang peroksidasi lipid membran sel dan penghancuran sel, sehingga porfiria sering disertai dengan fotosensitifitas dan ulserasi pada kulit yang terpapar.
Porfirinogen tidak berwarna dan tidak berfluoresensi, sedangkan porfirin menunjukkan fluoresensi merah yang intens di bawah sinar ultraviolet. Porfirin berlebih, yang diekskresikan dalam urin, memberinya warna gelap ("porfirin" dalam bahasa Yunani berarti ungu).
Kadang-kadang, dalam bentuk ringan porfiria herediter, penyakit ini mungkin tidak menunjukkan gejala, tetapi minum obat yang menginduksi sintesis aminolevulinate sintase dapat menyebabkan eksaserbasi penyakit. Dalam beberapa kasus, gejala penyakit tidak muncul sampai pubertas, ketika peningkatan pembentukan -steroid menyebabkan induksi sintesis aminolevulinate sintase. Porfiria juga diamati dalam kasus keracunan dengan garam timbal, karena timbal menghambat aminolevulinate dehidratase dan ferrochelatase. Beberapa herbisida dan insektisida yang mengandung halogen merupakan penginduksi sintesis aminolevulinat sintase, sehingga konsumsinya disertai dengan gejala porfiria.
Porfiria intermiten akut (API) - penyebabnya adalah cacat pada gen penyandi PBG - deaminase. Diwariskan secara autosomal dominan. Ada akumulasi prekursor awal sintesis heme: 5-ALA (5-ALA) dan porfobilinogen (PBG).
PBG yang tidak berwarna diubah menjadi porfibilin dan porfirin dalam cahaya, mereka memberikan warna gelap pada urin. ALA memiliki efek neurotoksik, menyebabkan kelumpuhan flaccid pada ekstremitas dan paresis otot pernapasan. Yang terakhir menyebabkan akut gagal napas. Penyakit ini memanifestasikan dirinya di usia paruh baya, dipicu oleh penggunaan analgesik, obat sulfanilamide, karena mereka meningkatkan sintesis ALA - sintase.
Gejala klinisnya adalah nyeri perut akut, muntah, konstipasi, gangguan kardiovaskular, gangguan neuropsikiatri. Tidak ada peningkatan kepekaan terhadap cahaya, karena gangguan metabolisme terjadi pada tahap sebelum pembentukan uroporfirinogen.
Untuk pengobatan, obat normosang - heme arginat digunakan. Tindakan ini didasarkan pada fakta bahwa heme, melalui mekanisme umpan balik negatif, memblok translasi ALA-sintase, dan, akibatnya, sintesis ALA dan PBG menurun, yang menghasilkan pengurangan gejala.
Porfiria eritropoietik kongenital adalah penyakit kongenital yang lebih langka yang diturunkan secara resesif autosomal. Sifat molekuler penyakit ini tidak diketahui secara pasti; telah ditetapkan, bagaimanapun, bahwa hal itu ditandai dengan ketidakseimbangan tertentu dalam aktivitas relatif uroporfirinogen-III-kosintase dan uroporfirinogen-1-sintase. Pembentukan uroporfirinogen I secara kuantitatif secara signifikan melebihi sintesis isomer normal uroporfirinogen III dalam jalur sintesis heme. Meskipun kelainan genetik meluas ke semua sel, itu memanifestasikan dirinya, untuk alasan yang tidak diketahui, terutama di jaringan eritropoietik. Pasien dengan porfiria eritropoietik kongenital mengekskresikan sejumlah besar isomer tipe I uroporfirinogen dan koproporfirinogen; dalam urin, kedua senyawa ini secara spontan teroksidasi menjadi uroporfirin I dan koproporfirin I, pigmen fluoresen merah. Sebuah kasus telah dilaporkan di mana ada sedikit peningkatan konsentrasi uroporfirin III, tetapi rasio isomer tipe I dan III kira-kira 100:1. Eritrosit yang bersirkulasi mengandung sejumlah besar uroporfirin 1, namun konsentrasi tertinggi porfirin ini tercatat dalam sel sumsum tulang (tetapi tidak pada hepatosit).
Sensitivitas kulit dicatat, karena sifat spektrum penyerapan senyawa porfirin, yang terbentuk dalam jumlah besar. Pasien memiliki retakan pada kulit, dan fenomena hemolitik sering diamati.
Koproporfiria herediter adalah kelainan dominan autosomal yang disebabkan oleh defisiensi koproporfirinogen oksidase, suatu enzim mitokondria yang bertanggung jawab atas konversi koproporfirinogen III menjadi protoporfirinogen IX. Koproporfirinogen III diekskresikan dalam jumlah besar dari tubuh dalam komposisi tinja, dan juga, karena kelarutannya dalam air, diekskresikan dalam dalam jumlah besar dengan urin. Seperti uroporfirinogen, koproporfirinogen dengan cepat teroksidasi dalam cahaya dan udara, berubah menjadi pigmen merah koproporfirin.
Kemampuan terbatas untuk mensintesis heme pada penyakit ini (terutama dalam kondisi stres) menyebabkan derepresi ALA-siitase. Akibatnya, terjadi pembentukan ALA dan porfobilinogen yang berlebihan, serta zat antara lain dalam jalan sintesis tema, yang terbentuk pada tahap sebelum tahap penghambatan herediter. Oleh karena itu, pasien dengan koproporfiria herediter menunjukkan semua tanda dan gejala yang berhubungan dengan kelebihan ALA dan porfobilinogen yang merupakan karakteristik dari porfiria akut intermiten, tetapi selain itu mereka juga mengalami peningkatan fotosensitifitas karena adanya jumlah koproporfirinogen dan uroporfirinogen yang berlebihan. Pada penyakit ini, pemberian hematin juga dapat menginduksi setidaknya represi parsial ALA sintase dan mengurangi gejala yang terkait dengan produksi intermediet biosintetik heme yang berlebihan.
Porfiria mosaik, atau fotoporfiria herediter, adalah kelainan dominan autosomal di mana ada penyumbatan parsial konversi enzimatik protoporfirinogen menjadi heme. Biasanya, transformasi ini dilakukan oleh dua enzim, protoporfirinogen oksidase dan ferrochelatase, yang terlokalisasi di mitokondria. Dilihat dari data yang diperoleh pada kultur fibroblas kulit, pada pasien dengan porfiria mosaik, kandungan protoporfirinogen oksidase hanya setengah dari jumlah normal. Pada pasien dengan porfiria mosaik, kandungan heme relatif berkurang dalam kondisi stres, serta keadaan ALA sintase hepatik yang tertekan. Seperti disebutkan di atas, peningkatan aktivitas ALA sintase menyebabkan kelebihan produksi semua zat antara sintesis heme di daerah sebelum tahap yang diblokir. Jadi, pasien dengan porfiria mosaik mengekskresikan ALA, porfobilinogen, uroporfirin, dan koproporfirin dalam jumlah berlebih dalam urin, dan mengekskresikan uroporfirin, koproporfirin, dan protoporfirin dalam feses. Urin pasien berpigmen dan berfluoresensi, dan kulit sensitif terhadap cahaya dengan cara yang sama seperti pada pasien dengan porfiria kutaneous tardive (lihat di bawah).
Porfiria tardio kulit mungkin merupakan bentuk porfiria yang paling umum. Biasanya dikaitkan dengan beberapa jenis kerusakan hati, terutama dengan konsumsi alkohol yang berlebihan atau kelebihan ion besi. Sifat gangguan metabolisme belum ditetapkan secara pasti, tetapi kemungkinan penyebab adalah defisiensi parsial uroporfirinogen dekarboksilase. Gangguan tersebut tampaknya ditransmisikan sebagai sifat dominan autosomal, tetapi penetrasi genetik bervariasi dan dalam banyak kasus tergantung pada adanya disfungsi hati. Seperti yang diperkirakan, urin mengandung peningkatan kadar uroporfirin tipe I dan III; pada saat yang sama, ekskresi urin ALA dan porfobilinogen relatif jarang. Terkadang urin mengandung porfirin dalam jumlah yang sangat signifikan, memberikan warna merah muda; ketika diasamkan, paling sering memberikan fluoresensi merah muda di wilayah ultraviolet.
Hati mengandung sejumlah besar porfirin dan oleh karena itu berfluoresensi kuat, sedangkan eritrosit dan sel sumsum tulang tidak berfluoresensi. Manifestasi klinis utama pada porfiria kutaneous tardive adalah peningkatan fotosensitifitas kulit. Pada pasien, tidak ada peningkatan aktivitas ALA sintase, atau, masing-masing, kelebihan kandungan porfobilinogen dan ALA dalam urin yang diamati; ini berkorelasi dengan tidak adanya karakteristik serangan akut porfiria akut intermiten.
Protoporfiria, atau protoporfiria eritropoietik, tampaknya disebabkan oleh defisiensi aktivitas ferrochelatase yang diturunkan secara dominan dalam mitokondria semua jaringan; Secara klinis, penyakit ini bermanifestasi sebagai urtikaria akut yang disebabkan oleh paparan sinar matahari. Sel darah merah, plasma, dan feses mengandung jumlah protoporfirin IX yang tinggi, dan retikulosit (sel darah merah yang belum matang) dan kulit (bila diperiksa dengan biopsi) sering berpendar dengan lampu merah. Hati mungkin juga berkontribusi terhadap peningkatan produksi protoporfirin IX, tetapi ekskresi urin porfirin dan prekursornya tidak diamati.
Biokimia Porfiria
Porfiria adalah sekelompok penyakit heterogen yang ditandai dengan peningkatan sekresi porfirin atau prekursornya. Beberapa bentuk porfiria bersifat herediter, yang lain didapat. Beberapa berbagai klasifikasi porfiria. Lebih mudah untuk membagi bentuk herediter menjadi tiga kelompok besar - eritropoietik, hati, dan bentuk semacam itu di mana gangguan metabolisme diamati secara bersamaan di jaringan eritropoietik dan hati (Tabel 33.2). Sebagian besar bentuk yang diturunkan dicirikan oleh adanya metabolisme
Tabel 33.2. Klasifikasi porfiria manusia
gangguan pribadi di semua jaringan, bagaimanapun, mereka muncul untuk beberapa alasan dalam satu jenis jaringan. Berikut ini adalah gambaran kelainan biokimia yang menjadi ciri porfiria.
Setiap jenis porfiria dicirikan oleh satu set porfirin dan prekursornya diekskresikan dalam urin. Data ini dan hubungannya dengan berbagai tahap sintesis heme ditunjukkan pada Gambar. 33.11.
Porfiria akut intermiten (POP) adalah ciri dari autosomal dominan penyakit keturunan seseorang, yang biasanya memanifestasikan dirinya hanya setelah mencapai pubertas. Penyebabnya adalah defisiensi parsial uroporfirinogen-1 sintase yang diturunkan. Pasien heterozigot untuk gen struktural yang rusak, sehingga aktivitas uroporfirinogen-1-sintase dalam sel mereka adalah 50% dari norma. Pasien dengan POP mengeluarkan sejumlah besar porfobilinogen dan ALA dalam urin. Kedua senyawa ini tidak berwarna, tetapi porfobilinogen dalam cahaya dan udara secara spontan membentuk dua produk berwarna - porfobilin dan porfirin. Ini adalah alasan untuk menggelapkan urin ketika berdiri di bawah cahaya dengan akses udara.
Porfobilinogen dan ALA terdapat dalam plasma dan cairan serebrospinal pasien, terutama selama eksaserbasi yang tajam. Obat-obatan dan hormon steroid yang metabolismenya memerlukan partisipasi protein yang mengandung heme, seperti sitokrom P-450, dapat mempercepat timbulnya eksaserbasi. Senyawa yang menginduksi porfiria selama metabolisme meningkatkan konsumsi protein heme dan dengan demikian mengurangi konsentrasi heme intraseluler; ini menyebabkan derepresi sintesis ALA sintase. Peningkatan aktivitas ALA sintase dan pemblokiran parsial uroporfirinogen-1 sintase menyebabkan akumulasi ALA dan porfobilinogen yang signifikan; itu disertai rasa sakit yang tajam di perut, muntah, sembelit, gangguan kardiovaskular, serta gangguan neuropsikiatri. Perlu dicatat bahwa, menurut data eksperimental, penurunan kandungan heme menghambat aktivitas triptofan pirolase dan menyebabkan akumulasi senyawa neuroaktif - triptofan dan 5-hidroksitriptamin.
Pasien dengan POP tidak mengalami peningkatan kepekaan terhadap cahaya, yang merupakan karakteristik dari porfiria hati lainnya. Hal ini diharapkan, karena baik porfirin maupun porfirinogen tidak terakumulasi pada pasien, karena gangguan metabolisme sintesis heme terjadi pada tahap sebelum pembentukan porfirinogen pertama (uroporfirinogen).
Seperti disebutkan di atas, gangguan metabolisme juga ditemukan di sel lain, khususnya di eritrosit, serta di fibroblas yang dikultur atau sel cairan ketuban; namun, peningkatan aktivitas ALA sintase, yang menyebabkan produksi berlebihan ALA dan porfobilinogen, terjadi terutama di hati. Rupanya, ini disebabkan oleh fakta bahwa hati adalah organ di mana metabolisme agen penginduksi terjadi. Porfiria akut adalah salah satu contoh penyakit langka yang secara fenotip diekspresikan pada heterozigot, sedangkan defisiensi enzim hanya 50%.
Seperti yang bisa diprediksi berdasarkan
Beras. 33.11. Tahap berurutan dari biosintesis heme, menunjukkan prekursor yang diekskresikan dalam urin, dengan berbagai bentuk porfiria Kurung keriting menggabungkan senyawa yang diekskresikan dalam urin dalam jumlah berlebih selama eksaserbasi bentuk porfiria ini. ALA - asam 5-aminolevulinat. (Direproduksi dengan modifikasi dari Kaufman L., Merver H. S. Cacat biokimia dalam dua jenis prfiria hati manusia. N. Engl. J. Med 1970:283:954.)
Menurut mekanisme yang diusulkan pengaturan sintesis ALA sintase oleh sistem represi - derepresin, pemberian hematin pada pasien dengan POP dapat mengurangi induksi ALA sintase dan dengan demikian meringankan perjalanan penyakit.
Porfiria erntropoietik kongenital adalah penyakit kongenital yang lebih langka yang diturunkan secara resesif autosomal. Sifat molekuler penyakit ini tidak diketahui secara pasti; telah ditetapkan, bagaimanapun, bahwa hal itu ditandai dengan ketidakseimbangan tertentu dalam aktivitas relatif uroporfirinogen-III-kosintase dan uroporfirinogen-1-sintase. Pembentukan uroporfirinogen I secara kuantitatif secara signifikan melebihi sintesis uroporfirinogen III, isomer normal dalam jalur sintesis heme. Meskipun kelainan genetik meluas ke semua sel, itu memanifestasikan dirinya, untuk alasan yang tidak diketahui, terutama di jaringan eritropoietik. Pasien dengan porfiria eritropoietik kongenital mengekskresikan sejumlah besar isomer tipe I uroporfirinogen dan koproporfirinogen; dalam urin, kedua senyawa ini secara spontan teroksidasi menjadi uroporfirin 1 dan koproporfirin I, pigmen fluoresen merah. Sebuah kasus telah dilaporkan di mana ada sedikit peningkatan konsentrasi uroporfirin III, tetapi rasio isomer tipe I dan III kira-kira 100:1. Eritrosit yang bersirkulasi mengandung sejumlah besar uroporfirin I, namun konsentrasi tertinggi porfirin ini tercatat dalam sel sumsum tulang (tetapi tidak pada hepatosit).
Mungkin karena pembentukan jumlah yang lebih kecil dari prekursor sejati heme, uroporfirinogen III, dan defisiensi relatif heme yang dihasilkan dalam jaringan eritropoietik pasien, ALA sintase diinduksi. Induksi ini menyebabkan kelebihan produksi porfirinogen tipe I. Seiring dengan peningkatan sintesis ALA sintase dan kelebihan produksi porfirinogen tipe I, pembentukan dan ekskresi porfobilinogen dan ALA meningkat. Jadi, berdasarkan penyimpangan biokimia, adalah mungkin untuk memprediksi penampilan gejala klinis, mirip dengan yang diamati pada POP, tetapi selain itu, fotosensitifitas kulit dicatat,
karena sifat spektrum serapan senyawa porfirin, yang terbentuk dalam jumlah banyak. Pasien memiliki retakan pada kulit, dan fenomena hemolitik sering diamati.
Coproporphyria herediter adalah kelainan autosomal dominan yang disebabkan oleh defisiensi coproporphyrinogen oxindase, enzim mitokondria yang bertanggung jawab untuk konversi coproporphyrinogen III menjadi protoporphyrinogen IX. Koproporfirinogen III diekskresikan dalam jumlah besar dari tubuh dalam tinja, dan karena kelarutannya dalam air, ia diekskresikan dalam jumlah besar dalam urin. Seperti uroporfirinogen, koproporfirinogen dengan cepat teroksidasi dalam cahaya dan udara, berubah menjadi pigmen merah koproporfirin.
Kemampuan terbatas untuk mensintesis heme pada penyakit ini (terutama dalam kondisi stres) menyebabkan derepresi ALA sintase. Akibatnya, terjadi kelebihan pembentukan ALA dan porfobilinogen, serta zat antara lain pada jalur sintesis heme, yang terbentuk pada tahap sebelum tahap penghambatan herediter. Oleh karena itu, pasien dengan koproporfiria herediter menunjukkan semua tanda dan gejala yang berhubungan dengan kelebihan ALA dan porfobilinogen yang merupakan karakteristik dari porfiria akut intermiten, tetapi selain itu mereka juga mengalami peningkatan fotosensitifitas karena adanya jumlah koproporfirinogen dan uroporfirinogen yang berlebihan. Pada penyakit ini, pemberian hematin juga dapat menginduksi setidaknya represi parsial ALA sintase dan mengurangi gejala yang terkait dengan produksi intermediet biosintetik heme yang berlebihan.
Porfiria mosaik, atau fotokoproporfiria herediter, adalah kelainan dominan autosomal di mana ada penyumbatan parsial konversi enzimatik protoporfirinogen menjadi heme. Biasanya, transformasi ini dilakukan oleh dua enzim, protoporfirinogen oksidase dan ferrochelatase, yang terlokalisasi di mitokondria. Dilihat dari data yang diperoleh pada kultur fibroblas kulit, pada pasien dengan porfiria mosaik, kandungan protoporfirinogen oksidase hanya setengah dari jumlah normal. Pada pasien dengan porfiria mosaik, kandungan heme relatif berkurang dalam kondisi stres, serta keadaan ALA sintase hepatik yang tertekan. Seperti disebutkan di atas, peningkatan aktivitas ALA sintase menyebabkan kelebihan produksi semua zat antara sintesis heme di daerah sebelum tahap yang diblokir. Jadi, pasien dengan porfiria mosaik mengekskresikan ALA, porfobilinogen, uroporfirin, dan koproporfirin dalam jumlah berlebih dalam urin, dan mengekskresikan uroporfirin, koproporfirin, dan protoporfirin dalam feses. Urin pasien berpigmen dan berfluoresensi, dan kulit sensitif terhadap cahaya dengan cara yang sama seperti pada pasien dengan porfiria kutaneous tardive (lihat di bawah).
Porfiria tardio kulit mungkin merupakan bentuk porfiria yang paling umum. Biasanya dikaitkan dengan beberapa jenis kerusakan hati, terutama dengan konsumsi alkohol yang berlebihan atau kelebihan ion besi. Sifat gangguan metabolisme belum ditetapkan secara pasti, tetapi kemungkinan penyebabnya adalah defisiensi parsial uroporphinogen decarboxylase. Gangguan tersebut tampaknya ditransmisikan sebagai sifat dominan autosomal, tetapi penetrasi genetik bervariasi dan dalam banyak kasus tergantung pada adanya disfungsi hati. Seperti yang diperkirakan, urin mengandung peningkatan kadar uroporfirin tipe I dan III; pada saat yang sama, ekskresi urin ALA dan porfobilinogen relatif jarang. Terkadang urin mengandung porfirin dalam jumlah yang sangat signifikan, memberikan warna merah muda; ketika diasamkan, paling sering memberikan fluoresensi merah muda di wilayah ultraviolet.
Hati mengandung sejumlah besar porfirin dan oleh karena itu berfluoresensi kuat, sedangkan eritrosit dan sel sumsum tulang tidak berfluoresensi. Ketua Manifestasi klinis dengan porfiria kulit lanjut adalah peningkatan fotosensitifitas kulit. Pada pasien, tidak ada peningkatan aktivitas ALA sintase, atau, masing-masing, kelebihan kandungan porfobilinogen dan ALA dalam urin yang diamati; ini berkorelasi dengan tidak adanya karakteristik serangan akut porfiria akut intermiten.
Protoporfiria, atau protoporfiria eritropoietik, tampaknya disebabkan oleh defisiensi aktivitas ferrochelatase yang diturunkan secara dominan dalam mitokondria semua jaringan; Secara klinis, penyakit ini bermanifestasi sebagai urtikaria akut yang disebabkan oleh paparan sinar matahari. Sel darah merah, plasma, dan feses mengandung jumlah protoporfirin IX yang tinggi, dan retikulosit (sel darah merah yang belum matang) dan kulit (bila diperiksa dengan biopsi) sering berpendar dengan lampu merah.
Hati mungkin juga berkontribusi terhadap peningkatan produksi protoporfirin IX, tetapi ekskresi urin porfirin dan prekursornya tidak diamati.
Porfiria didapat (beracun) dapat disebabkan oleh aksi senyawa beracun seperti heksaklorobenzena, garam timbal dan logam berat lainnya, serta obat-obatan seperti griseofulvin. Logam berat merupakan penghambat sejumlah enzim dalam sistem sintesis heme, termasuk ALA dehidratase, uroporfirinogen sintase, dan ferrochelatase.
Bagian I. Struktur dan fungsi protein dan enzim
Bagian II. Bioenergi dan metabolisme karbohidrat dan lipid
Bagian III. Metabolisme protein dan asam amino
Menyalin informasi dari halaman hanya diperbolehkan dengan tautan ke situs ini
Porfiria adalah sekelompok penyakit heterogen yang ditandai dengan peningkatan sekresi porfirin atau prekursornya. Beberapa bentuk porfiria bersifat herediter, yang lain didapat. Beberapa klasifikasi yang berbeda dari porfiria telah diusulkan. Lebih mudah untuk membagi bentuk herediter menjadi tiga kelompok besar - eritropoietik, hati, dan bentuk semacam itu di mana gangguan metabolisme diamati secara bersamaan di jaringan eritropoietik dan hati (Tabel 33.2). Sebagian besar bentuk yang diturunkan dicirikan oleh adanya metabolisme
Tabel 33.2. Klasifikasi porfiria manusia
gangguan pribadi di semua jaringan, bagaimanapun, mereka muncul untuk beberapa alasan dalam satu jenis jaringan. Berikut ini adalah gambaran kelainan biokimia yang menjadi ciri porfiria.
Setiap jenis porfiria dicirikan oleh satu set porfirin dan prekursornya diekskresikan dalam urin. Data ini dan hubungannya dengan berbagai tahap sintesis heme ditunjukkan pada Gambar. 33.11.
Porfiria akut intermiten (POP) adalah tanda penyakit keturunan autosomal dominan pada seseorang, yang biasanya tidak muncul sampai setelah pubertas. Penyebabnya adalah defisiensi parsial uroporfirinogen-1 sintase yang diturunkan. Pasien heterozigot untuk gen struktural yang rusak, sehingga aktivitas uroporfirinogen-1-sintase dalam sel mereka adalah 50% dari norma. Pasien dengan POP mengeluarkan sejumlah besar porfobilinogen dan ALA dalam urin. Kedua senyawa ini tidak berwarna, tetapi porfobilinogen dalam cahaya dan udara secara spontan membentuk dua produk berwarna - porfobilin dan porfirin. Ini adalah alasan untuk menggelapkan urin ketika berdiri di bawah cahaya dengan akses udara.
Porfobilinogen dan ALA terdapat dalam plasma dan cairan serebrospinal pasien, terutama selama eksaserbasi yang tajam. Obat-obatan dan hormon steroid yang metabolismenya memerlukan partisipasi protein yang mengandung heme, seperti sitokrom P-450, dapat mempercepat timbulnya eksaserbasi. Senyawa yang menginduksi porfiria selama metabolisme meningkatkan konsumsi protein heme dan dengan demikian mengurangi konsentrasi intraseluler heme; ini menyebabkan derepresi sintesis ALA sintase. Peningkatan aktivitas ALA sintase dan pemblokiran parsial uroporfirinogen-1 sintase menyebabkan akumulasi ALA dan porfobilinogen yang signifikan; ini disertai dengan sakit perut akut, muntah, sembelit, gangguan kardiovaskular, serta gangguan neuropsikiatri. Perlu dicatat bahwa, menurut data eksperimental, penurunan kandungan heme menghambat aktivitas triptofan pirolase dan menyebabkan akumulasi senyawa neuroaktif - triptofan dan 5-hidroksitriptamin.
Pasien dengan POP tidak mengalami peningkatan kepekaan terhadap cahaya, yang merupakan karakteristik dari porfiria hati lainnya. Hal ini diharapkan, karena baik porfirin maupun porfirinogen tidak terakumulasi pada pasien, karena gangguan metabolisme sintesis heme terjadi pada tahap sebelum pembentukan porfirinogen pertama (uroporfirinogen).
Seperti disebutkan di atas, gangguan metabolisme juga ditemukan di sel lain, khususnya di eritrosit, serta di fibroblas yang dikultur atau sel cairan ketuban; namun, peningkatan aktivitas ALA sintase, yang menyebabkan produksi berlebihan ALA dan porfobilinogen, terjadi terutama di hati. Rupanya, ini disebabkan oleh fakta bahwa hati adalah organ di mana metabolisme agen penginduksi terjadi. Porfiria akut adalah salah satu contoh penyakit langka yang secara fenotip diekspresikan pada heterozigot, sedangkan defisiensi enzim hanya 50%.
Seperti yang bisa diprediksi berdasarkan

Beras. 33.11. Tahapan berurutan dari biosintesis heme, menunjukkan prekursor yang diekskresikan dalam urin, dalam berbagai bentuk porfiria Kurung keriting menggabungkan senyawa yang diekskresikan dalam urin dalam jumlah berlebih selama eksaserbasi bentuk porfiria ini. ALA - asam 5-aminolevulinat. (Direproduksi dengan modifikasi dari Kaufman L., Merver H. S. Cacat biokimia dalam dua jenis prfiria hati manusia. N. Engl. J. Med 1970:283:954.)
Menurut mekanisme yang diusulkan pengaturan sintesis ALA sintase oleh sistem represi - derepresin, pemberian hematin pada pasien dengan POP dapat mengurangi induksi ALA sintase dan dengan demikian meringankan perjalanan penyakit.
Porfiria erntropoietik kongenital adalah penyakit kongenital yang lebih langka yang diturunkan secara resesif autosomal. Sifat molekuler penyakit ini tidak diketahui secara pasti; telah ditetapkan, bagaimanapun, bahwa hal itu ditandai dengan ketidakseimbangan tertentu dalam aktivitas relatif uroporfirinogen-III-kosintase dan uroporfirinogen-1-sintase. Pembentukan uroporfirinogen I secara kuantitatif secara signifikan melebihi sintesis uroporfirinogen III, isomer normal dalam jalur sintesis heme. Meskipun kelainan genetik meluas ke semua sel, itu memanifestasikan dirinya, untuk alasan yang tidak diketahui, terutama di jaringan eritropoietik. Pasien dengan porfiria eritropoietik kongenital mengekskresikan sejumlah besar isomer tipe I uroporfirinogen dan koproporfirinogen; dalam urin, kedua senyawa ini secara spontan teroksidasi menjadi uroporfirin 1 dan koproporfirin I, pigmen fluoresen merah. Sebuah kasus telah dilaporkan di mana ada sedikit peningkatan konsentrasi uroporfirin III, tetapi rasio isomer tipe I dan III kira-kira 100:1. Eritrosit yang bersirkulasi mengandung sejumlah besar uroporfirin I, tetapi konsentrasi tertinggi porfirin ini terdapat pada sel sumsum tulang (tetapi tidak pada hepatosit).
Mungkin karena pembentukan jumlah yang lebih kecil dari prekursor sejati heme, uroporfirinogen III, dan defisiensi relatif heme yang dihasilkan dalam jaringan eritropoietik pasien, ALA sintase diinduksi. Induksi ini menyebabkan kelebihan produksi porfirinogen tipe I. Seiring dengan peningkatan sintesis ALA sintase dan kelebihan produksi porfirinogen tipe I, pembentukan dan ekskresi porfobilinogen dan ALA meningkat. Jadi, berdasarkan kelainan biokimia, adalah mungkin untuk memprediksi munculnya gejala klinis yang serupa dengan yang diamati pada POP, tetapi selain itu, fotosensitifitas kulit dicatat,
karena sifat spektrum serapan senyawa porfirin, yang terbentuk dalam jumlah banyak. Pasien memiliki retakan pada kulit, dan fenomena hemolitik sering diamati.
Coproporphyria herediter adalah kelainan autosomal dominan yang disebabkan oleh defisiensi coproporphyrinogen oxindase, enzim mitokondria yang bertanggung jawab untuk konversi coproporphyrinogen III menjadi protoporphyrinogen IX. Koproporfirinogen III diekskresikan dalam jumlah besar dari tubuh dalam tinja, dan karena kelarutannya dalam air, ia diekskresikan dalam jumlah besar dalam urin. Seperti uroporfirinogen, koproporfirinogen dengan cepat teroksidasi dalam cahaya dan udara, berubah menjadi pigmen merah koproporfirin.
Kemampuan terbatas untuk mensintesis heme pada penyakit ini (terutama dalam kondisi stres) menyebabkan derepresi ALA sintase. Akibatnya, terjadi kelebihan pembentukan ALA dan porfobilinogen, serta zat antara lain pada jalur sintesis heme, yang terbentuk pada tahap sebelum tahap penghambatan herediter. Oleh karena itu, pasien dengan koproporfiria herediter menunjukkan semua tanda dan gejala yang berhubungan dengan kelebihan ALA dan porfobilinogen yang merupakan karakteristik dari porfiria akut intermiten, tetapi selain itu mereka juga mengalami peningkatan fotosensitifitas karena adanya jumlah koproporfirinogen dan uroporfirinogen yang berlebihan. Pada penyakit ini, pemberian hematin juga dapat menginduksi setidaknya represi parsial ALA sintase dan mengurangi gejala yang terkait dengan produksi intermediet biosintetik heme yang berlebihan.
Porfiria mosaik, atau fotokoproporfiria herediter, adalah kelainan dominan autosomal di mana ada penyumbatan parsial konversi enzimatik protoporfirinogen menjadi heme. Biasanya, transformasi ini dilakukan oleh dua enzim, protoporfirinogen oksidase dan ferrochelatase, yang terlokalisasi di mitokondria. Dilihat dari data yang diperoleh pada kultur fibroblas kulit, pada pasien dengan porfiria mosaik, kandungan protoporfirinogen oksidase hanya setengah dari jumlah normal. Pada pasien dengan porfiria mosaik, kandungan heme relatif berkurang dalam kondisi stres, serta keadaan ALA sintase hepatik yang tertekan. Seperti disebutkan di atas, peningkatan aktivitas ALA sintase menyebabkan kelebihan produksi semua zat antara sintesis heme di daerah sebelum tahap yang diblokir. Jadi, pasien dengan porfiria mosaik mengekskresikan ALA, porfobilinogen, uroporfirin, dan koproporfirin dalam jumlah berlebih dalam urin, dan mengekskresikan uroporfirin, koproporfirin, dan protoporfirin dalam feses. Urin pasien berpigmen dan berfluoresensi, dan kulit sensitif terhadap cahaya dengan cara yang sama seperti pada pasien dengan porfiria kutaneous tardive (lihat di bawah).
Porfiria tardio kulit mungkin merupakan bentuk porfiria yang paling umum. Biasanya dikaitkan dengan beberapa jenis kerusakan hati, terutama dengan konsumsi alkohol yang berlebihan atau kelebihan ion besi. Sifat gangguan metabolisme belum ditetapkan secara pasti, tetapi kemungkinan penyebabnya adalah defisiensi parsial uroporphinogen decarboxylase. Gangguan tersebut tampaknya ditransmisikan sebagai sifat dominan autosomal, tetapi penetrasi genetik bervariasi dan dalam banyak kasus tergantung pada adanya disfungsi hati. Seperti yang diperkirakan, urin mengandung peningkatan kadar uroporfirin tipe I dan III; pada saat yang sama, ekskresi urin ALA dan porfobilinogen relatif jarang. Terkadang urin mengandung porfirin dalam jumlah yang sangat signifikan, memberikan warna merah muda; ketika diasamkan, paling sering memberikan fluoresensi merah muda di wilayah ultraviolet.
Hati mengandung sejumlah besar porfirin dan karena itu berfluoresensi kuat, sedangkan eritrosit dan sel sumsum tulang tidak. Manifestasi klinis utama pada porfiria kutaneous tardive adalah peningkatan fotosensitifitas kulit. Pada pasien, tidak ada peningkatan aktivitas ALA sintase, atau, masing-masing, kelebihan kandungan porfobilinogen dan ALA dalam urin yang diamati; ini berkorelasi dengan tidak adanya karakteristik serangan akut porfiria akut intermiten.
Protoporfiria, atau protoporfiria eritropoietik, tampaknya disebabkan oleh defisiensi aktivitas ferrochelatase yang diturunkan secara dominan dalam mitokondria semua jaringan; Secara klinis, penyakit ini bermanifestasi sebagai urtikaria akut yang disebabkan oleh paparan sinar matahari. Sel darah merah, plasma, dan feses mengandung jumlah protoporfirin IX yang tinggi, dan retikulosit (sel darah merah yang belum matang) dan kulit (bila diperiksa dengan biopsi) sering berpendar dengan lampu merah.
Hati mungkin juga berkontribusi terhadap peningkatan produksi protoporfirin IX, tetapi ekskresi urin porfirin dan prekursornya tidak diamati.
Porfiria didapat (beracun) dapat disebabkan oleh aksi senyawa beracun seperti heksaklorobenzena, garam timbal dan logam berat lainnya, serta obat-obatan seperti griseofulvin. Logam berat merupakan penghambat sejumlah enzim dalam sistem sintesis heme, termasuk ALA dehidratase, uroporfirinogen sintase, dan ferrochelatase.
Urs A. Meyer
Porfiria adalah patologi yang terkait dengan kelainan herediter atau didapat dalam biosintesis heme. Porfirin, pigmen tetrapirol ini, bertindak sebagai perantara dalam jalur ini dan terbentuk dari prekursor - 8 -asam aminolevulinat (ALA) dan porfobilinogen. Heme, kompleks besi besi dengan protoporfirin IX, berfungsi sebagai gugus prostetik hemoprotein seperti hemoglobin, sitokrom, katalase, dan triptofan oksigenase. Biosintesisnya sangat penting dan terjadi di semua sel aerobik.
Setiap porfiria dicirikan oleh fitur hiperproduksi, akumulasi dan ekskresi produk antara biosintesis heme. Fitur-fitur ini mencerminkan ekspresi metabolik dari defisiensi enzim biosintetik individu.
Manifestasi klinis utama adalah serangan intermiten disfungsi sistem saraf dan/atau sensitivitas kulit terhadap sinar matahari. Sindrom neurologis biasanya dipicu oleh penggunaan obat-obatan seperti barbiturat dan terdiri dari sakit perut, neuropati perifer, dan gangguan mental. Gejala neuropsik hanya muncul pada porfiria, di mana produksi prekursor porfirin - ALA dan porfobilinogen - meningkat tajam. Patogenesis gangguan neurologis tidak jelas. Sensitivitas cahaya pada kulit secara langsung berhubungan dengan peningkatan akumulasi porfirin, meskipun dengan berbagai pelanggaran manifestasi kulit tidak sama. Sensitivitas cahaya disebabkan oleh aksi fotodinamik porfirin dan mungkin dimediasi oleh oksigen singlet yang dihasilkan dengan perkembangan selanjutnya dari proses destruktif, seperti peroksidasi lipid membran lisosom. Porfiria manusia yang diturunkan secara dominan diekspresikan secara berbeda. Hanya perubahan biokimia atau enzimatik yang dapat ditentukan. Perjalanan penyakit laten seperti itu mungkin merupakan salah satu tahap atau berlanjut sepanjang hidup pasien. Dalam kasus lain, gejala dapat dipicu oleh obat-obatan, hormon, atau kerusakan hati.
Klasifikasi. Porfiria biasanya dibagi menjadi dua kelompok utama (eritropoietik dan hati) sesuai dengan situs utama sintesis heme, di mana "kesalahan" metabolisme dimanifestasikan. Satu-satunya bentuk porfiria eritropoietik murni, eritropoietik kongenital (CEP), jarang terjadi. Pada protoporfiria (PrP), porfirin terakumulasi baik di sel eritroid maupun di jaringan hati. Pada porfiria akut intermiten, koproporfiria herediter, dan porfiria beraneka ragam (masing-masing TIO, NCP, dan PP), defisiensi enzim yang diturunkan secara dominan menyebabkan pelanggaran biosintesis heme, terutama di hati, tanpa gangguan yang terlihat dalam pembentukan hemoglobin. Porfiria kutaneous kronis (CCP) sebelumnya dianggap sebagai penyakit hati yang didapat. Namun, sebagian besar (jika tidak semua) pasien memiliki defisiensi uroporfirinogen dekarboksilase herediter. Porfiria didapat, menyerupai HCP, disebabkan oleh paparan hidrokarbon poliklorin dan tumor hati. Keracunan timbal juga disertai dengan gangguan sintesis porfirin dan heme. Beberapa peningkatan ekskresi porfirin urin atau prekursornya, serta akumulasi porfirin dalam eritrosit, dapat menyertai banyak kondisi klinis. Dengan fenomena sekunder, tidak ada gejala dan tanda-tanda porfiria.
Aspek biokimia. Urutan reaksi sintesis heme dari substrat glisin dan enzim suksinil A melalui ALA dan porfobilinogen (PBA) dikatalisis oleh empat mitokondria dan empat enzim sitosol. Ada perbedaan dalam regulasi biosintesis heme di jaringan yang berbeda.
Di hati, laju pembentukan heme dibatasi oleh reaksi yang dikatalisis oleh ALA sintase. Enzim yang bekerja setelah ALA sintase ditemukan secara berlebihan. Pengatur utama ALA sintase adalah produk akhir dari seluruh jalur, heme, yang menekan enzim melalui mekanisme umpan balik negatif. Peningkatan kebutuhan heme dipenuhi oleh neoplasma ALA sintase. Sintesisnya di hati diinduksi oleh sejumlah besar zat yang larut dalam lemak, steroid dan senyawa kimia yang berfungsi sebagai substrat dan penginduksi hemoprotein sitokrom P 450 - oksidase akhir pada jalur metabolisme mikrosomal agen farmakologis. Induksi ini dimodulasi oleh banyak faktor genetik, metabolik dan lingkungan. Pada porfiria, di mana gejalanya dipicu oleh obat-obatan tertentu, saling ketergantungan sintesis heme dan oksidasi mikrosomal obat-obatan ini menjadi sangat penting.
Dalam sel sumsum tulang di mana sintesis heme lengkap terjadi, reaksi pembatas laju juga dikatalisis oleh ALA sintase, tetapi sedikit yang diketahui tentang perannya dalam sintesis heme selama pembelahan, diferensiasi, dan pematangan sel eritroid. Dalam proses pematangan sel-sel ini, inti dan mitokondria dan, akibatnya, enzim mitokondria menghilang dari mereka.sintesis heme, sedangkan enzim sitosol yang mengkatalisis reaksi antara ALK dan koproporfirinogen dipertahankan. Dalam hal ini, eritrosit dapat digunakan untuk mendiagnosis porfiria yang berhubungan dengan defek pada enzim sitosol saja.
Pengaturan sintesis heme di sumsum tulang dan hati berbeda. Di hati, penentu utama pembentukan heme adalah tingkat ALA sintase, sedangkan di sumsum tulang, sintesis heme dipicu oleh proses kompleks diferensiasi sel eritroid. Itulah sebabnya, mungkin, cacat pada enzim sintesis heme dalam sel eritroid dan hati memanifestasikan dirinya secara berbeda.
Porfirinogen menempati posisi perantara antara porfobilinogen dan protoporfirin. Mereka tidak berwarna dan tidak berpendar. Dengan pengecualian protoporfirin, porfirin adalah produk sampingan yang meninggalkan jalur biosintetik karena oksidasi ireversibel dari porfirinogen yang sesuai. Porfirin tidak melakukan fungsi fisiologis, tetapi berdasarkan warna dan fluoresensinya menentukan warna yang tidak biasa urin dan eritrosit pada beberapa pasien.
Lokasi dua rantai samping tersubstitusi pada cincin pirol porfirin menentukan tipe struktural isomer, yang diberi nomor dari I hingga IV. Hanya tipe I dan III yang ditemukan di alam, dan hanya tipe III yang berfungsi sebagai substrat untuk tahap akhir reaksi yang mengarah pada pembentukan protoporfirin IX dan heme. Selama pemecahan heme, bukan porfirin yang terbentuk, tetapi tetrapirol non-siklik, yang disebut pigmen empedu.
Porfiria eritropoietik kongenital
Definisi.Porfiria eritropoietik kongenital (CEP, penyakit Günther, porfiria fotosensitif kongenital, uroporfiria eritropoietik) adalah penyakit yang diturunkan secara resesif langka yang bermanifestasi sebagai fotosensitifitas kronis dengan lesi kulit yang merusak dan anemia hemolitik.
Genetika, frekuensi dan patogenesis.Pasien homozigot untuk gen resesif autosomal. Pada heterozigot, metabolisme porfirin jarang terganggu, secara lahiriah mereka terlihat sehat. Defek enzim yang mendasari belum ditetapkan, tetapi mungkin merupakan ketidakseimbangan fungsional antara aktivitas porfobilinogen deaminase dan uroporfirinogen cosynthase. AKU AKU AKU . Anomali ini diekspresikan secara eksklusif dalam sel eritroid yang matang dan menyebabkan hiperproduksi uroporfirinogen yang tajam. saya , sedangkan produksi uroporfirinogen AKU AKU AKU tidak berubah atau sedikit meningkat. Uroporfirinogen saya tidak dapat digunakan untuk sintesis heme, tetapi diubah menjadi koproporfirinogen SAYA. Uroporfirin I , koproporfirinogen saya dan koproporfirin saya menumpuk di jaringan dan diekskresikan secara berlebihan dalam urin dan feses.
Manifestasi klinis dan diagnosis.Pada pasien, porfirin menumpuk bahkan selama perkembangan janin. Sudah pada atau segera setelah melahirkan, urin merah muda atau merah biasanya mulai terpisah, sementara fotosensitifitas kulit, hemolisis periodik dan splenomegali dapat muncul kemudian. Hipertrikosis dan pewarnaan merah pada gigi dan tulang sering diidentifikasi. Kematian sudah bisa terjadi di masa kanak-kanak. Dengan kelangsungan hidup yang lebih lama, pasien mengembangkan bekas luka besar yang melumpuhkan, terutama pada kulit jari, hidung dan telinga. Sejumlah besar uroporfirin I, koproporfirin dan porfirin dengan gugus karboksil 7, 6, 5 dan 3 ditentukan dalam urin, sedangkan ekskresi AL C dan PBG tidak berubah. Sejumlah besar koproporfirin I ditemukan dalam tinja.Normoblas, retikulosit dan eritrosit mengandung sejumlah besar uroporfirin saya dan sejumlah kecil koproporfirinogen saya . Normoblas dan retikulosit menunjukkan fluoresensi merah yang intens. Sesuai dengan ekskresi normal AL C dan PBG, tidak ada patologi neurologis.
Perlakuan. Hindari paparan sinar matahari. Dalam beberapa kasus, anemia hemolitik, ekskresi porfirin, dan penurunan fotosensitifitas setelah splenektomi. Penggunaan infus hematin dan pemberian oral R-karoten belum melampaui ruang lingkup percobaan.
Porfiria hati
Tiga porfiria hati (IOP, NCP, dan PP) serupa dalam banyak hal. Semuanyamereka diwariskan sebagai sifat dominan autosomal. Serangan akut patologi neurologis yang mengancam jiwa dipicu oleh berbagai obat, hormon, dan faktor lain, di mana sejumlah besar ALA dan PBG diekskresikan dalam urin, tetapi jenis porfirin dalam urin dan feses berbeda.
Porfiria akut intermiten.Definisi. Porfiria akut intermiten [IOP, porfiria intermiten akut (AIP), piroloporfiria] ditandai dengan serangan berulang dari gejala neurologis dan psikiatri. Tidak ada sensitivitas cahaya. Fungsi porphobilinogen deaminase terutama terganggu.
Genetika, frekuensi dan patogenesis. Anomali ini diwariskan sebagai sifat dominan autosomal dengan ekspresivitas variabel. Gen abnormal terjadi pada frekuensi 1:10.000-1:50.000, tetapi di beberapa daerah mungkin lebih tinggi. Homozigot tidak ditemukan. Penyebab penyakit ini adalah defisiensi parsial (50%) porfobilinogen deaminase, yang mengubah PBG menjadi uroporfirinogen I. Pada tingkat gen, defisiensi ini dapat disebabkan oleh lebih dari satu mekanisme, tetapi mutasi yang paling umum menyebabkan penurunan jumlah enzim imunoreaktif protein. Di hati, defisiensi parsial enzim menyebabkan peningkatan aktivitas dan / atau induksibilitas ALA sintase. obat dan faktor-faktor lain dan karenanya meningkatkan pembentukan dan ekskresi urin dari ALA dan PBG. Dalam kondisi ini, porfirin tidak menumpuk, dan oleh karena itu fotosensitifitas kulit tidak meningkat. Pada TIO, aktivitas deaminase porfobilinogen yang berkurang terdeteksi di hati, eritrosit, kultur fibroblas kulit, leukosit, dan sel cairan ketuban. Dengan demikian, defek enzim juga terjadi pada jaringan ekstrahepatik, tetapi konsekuensi metaboliknya tidak dimanifestasikan di dalamnya. Defisiensi enzim tanpa adanya faktor yang didapat tidak serta merta menyebabkan porfiria akut yang bermakna secara klinis, dan hanya sepertiga pasien atau bahkan kurang dengan defek genetik ini pernah mengalami serangan porfiria.Hubungan antara defek genetik dan gangguan neurologis masih belum jelas. .
Manifestasi klinis dan diagnosis. Gejala penyakit jarang muncul sebelum pubertas. Biasanya gejala pertama dan paling mencolok dari serangan porfiria adalah sakit perut. Ini bisa menjadi sedang atau sangat kuat, kolik, lokal atau umum, menyebar ke punggung atau punggung bawah. Rasa sakit mungkin terkait dengan neuropati otonom, disertai dengan pelanggaran aktivitas motorik saluran pencernaan dengan bagian usus yang spasmodik dan melebar. Perut biasanya lunak dan rasa sakit tidak diperburuk oleh tekanan. Karena demam dan leukositosis yang sering dikaitkan, serangan akut porfiria dapat menyerupai apa pun proses inflamasi dalam rongga perut. Muntah parah dan konstipasi sering terjadi. Anomali neurologis dan mental memanifestasikan dirinya dengan cara yang berbeda. Fungsi saraf perifer, sistem saraf otonom, batang otak, saraf kranial, atau otak mungkin terganggu. Takikardia dan hipertensi labil dengan hipotensi postural, retensi urin, dan keringat berlebihan sering terjadi. Hipertensi dan takikardia berkorelasi dengan peningkatan ekskresi katekolamin. Neuropati perifer disebabkan oleh keterlibatan saraf motorik yang dominan dalam proses tersebut, tetapi komponen sensitif juga dapat bergabung. Refleks tendon dalam berkurang atau tidak ada. Nyeri neuritik yang khas pada ekstremitas, area hipo dan parestesia, serta kaki dan tangan yang lemas dan terkulai. Paraplegia atau quadriplegia lembek lengkap dapat terjadi. Di masa lalu, kelumpuhan otot-otot pernapasan menjadi penyebab utama kematian. Ketika saraf kranial terlibat dalam proses tersebut, saraf optik dapat mengalami atrofi, oftalmoplegia dan disfagia dapat bergabung. Dengan kerusakan yang lebih parah pada sistem saraf pusat, delirium, koma, dan kejang muncul. Meskipun neuropati reversibel, paresis residual dapat bertahan selama bertahun-tahun setelah serangan akut. Banyak pasien untuk waktu yang lama tetap mudah tersinggung, tidak stabil secara emosional dengan gangguan fungsional yang persisten. Pada 1/3 pasien, jiwa terganggu, sindrom otak organik dengan kecemasan, disorientasi, dan halusinasi visual dapat berkembang. Terkadang hiponatremia berat ditentukan. Hal ini mungkin disebabkan oleh beberapa penyebab (termasuk ekskresi natrium melalui saluran pencernaan, asupan cairan yang tidak tepat, dan bentuk nefropati pemborosan garam karena efek toksik ALA), tetapi yang utama tampaknya adalah sekresi hormon antidiuretik yang tidak memadai. . Dalam beberapa kasus, hipomagnesemia yang jelas bergabung sehingga tetani berkembang.
Serangan akut berlangsung selama berhari-hari atau bahkan berbulan-bulan dan bervariasi dalam frekuensi dan tingkat keparahan. Selama periode remisi, gejala penyakit melemah atau hilang sama sekali. Manifestasi klinis (dan biokimia) dapat dipicu oleh dosis konvensional (terapeutik) barbiturat, antikonvulsan, estrogen, kontrasepsi, atau alkohol. Semua zat ini dioksidasi oleh hemoprotein dari sistem sitokrom P450. Selama serangan akut, metabolisme beberapa di antaranya di hati mungkin terganggu. Pada beberapa wanita, kemunduran berkorelasi dengan siklus menstruasi, dan porfiria laten dapat menjadi jelas selama tanggal terlambat kehamilan atau segera setelah melahirkan. Kejang juga dapat dipicu oleh pengurangan asupan kalori (puasa) dalam waktu lama.
data laboratorium. Serangan akut ditandai dengan ekskresi urin yang berlebihan dari ALA dan PBG, dan atas dasar ini, TIO tidak berbeda dari NCP atau PP. Tingkat ALA dan PBG dalam urin tidak berkorelasi dengan tingkat keparahan gejala. Tes skrining yang sederhana dan andal untuk membantu mendiagnosis serangan akut TIO, LCP, dan PP adalah penentuan kualitatif porfobilinogen urin (tes Watson-Schwartz atau Hosh). Pada gangguan neuropsikiatri, tes ini hampir selalu positif, tetapi ini mengharuskan konsentrasi PBG dalam urin melebihi batas atas normal sebanyak 3-5 kali. Dalam hal ini, baik tes dalam bentuk laten penyakit atau normalisasi ekskresi PBG setelah serangan bisa menjadi negatif. Terkadang diperlukan kuantisasi diekskresikan dalam urin ALA dan PBG menggunakan metode kromatografi. Dalam bentuk laten TIO dengan ekskresi normal ALA dan PBG, diagnosis dapat ditegakkan berdasarkan hasil penentuan aktivitas porfobilinogen deaminase dalam eritrosit, leukosit, atau fibroblas kulit yang dikultur. Namun, pada pasien sehat dan TIO, hasil ini tumpang tindih, dan tidak selalu memungkinkan untuk membuat diagnosis yang akurat.
Dengan TIO, sesuai dengan defek enzim, ekskresi prekursor porfirin - ALA dan PBG meningkat, oleh karena itu, urin yang baru diperoleh biasanya tidak berwarna, mengandung sedikit uro- atau koproporfirin yang telah dibentuk sebelumnya. Ini dapat menjadi gelap saat berdiri, karena PBG secara spontan berpolimerisasi menjadi uroporfirin dan porfobilin, pigmen coklat tua dengan struktur yang tidak diketahui. Namun, pada beberapa pasien, pigmen yang terbentuk secara non-enzimatik dalam jumlah yang cukup ditentukan untuk memberikan warna merah tua pada urin yang baru diperoleh. Konsentrasi porfirin dalam tinja biasanya dalam kisaran normal.
Tes fungsi hati konvensional tidak berubah, dengan pengecualian peningkatan retensi bromsulfalein. Dalam darah tepi, massa eritrosit agak berkurang, volume darah juga berkurang, atau anemia normokromik normositik transien dicatat. Perubahan metabolik selama serangan akut termasuk hiperkolesterolemia yang berhubungan dengan peningkatan lipoprotein densitas rendah, peningkatan tiroksin serum (tanpa hipertiroidisme), gangguan toleransi glukosa, dan pemulihan 5a testosteron di hati. Hubungan anomali ini dengan cacat genetik masih belum jelas.
Perlakuan. Pengobatan selama serangan akut pada TIO, NCP dan PP adalah sama. Beberapa serangan akut tampaknya dapat diatasi dengan karbohidrat dalam jumlah besar (500 g/hari) (efek glukosa), meskipun belum ada studi objektif tentang efektivitas pengobatan ini. Direkomendasikan pemberian glukosa intravena dengan kecepatan 20 g/jam. Jika kondisi pasien tidak membaik setelah 48 jam pemberian glukosa terus menerus atau jika gejala neuropsikis berlanjut, hematin harus diberikan secara intravena (4 mg / kg selama 10-15 menit setiap 12 jam selama 3-6 hari). Ini tersedia secara komersial (pangematin) sebagai bubuk lyophilized. Solusi disiapkan segera sebelum infus. Ketika hematin digunakan pada dosis yang dianjurkan, komplikasi sangat jarang terjadi. Tromboflebitis tempat infus, koagulopati (dimanifestasikan oleh trombositopenia, pemanjangan waktu protrombin, beberapa perubahan waktu tromboplastin, dan hipofibrinogenemia) dan hemolisis kadang-kadang dilaporkan. Baik hematin dan glukosa pada hewan percobaan melawan induksi ALA sintase hati dan dalam waktu 48 jam dapat menetralkan perubahan biokimia dan menyebabkan perbaikan kondisi pasien. Untuk mencegah dan / atau memperbaiki hiponatri-, hipomagnesium- dan azotemia, penting untuk melakukan perawatan pemeliharaan dengan pemantauan yang cermat terhadap keadaan metabolisme air dan elektrolit. Takikardia dan hipertensi harus diobati dengan beta-blocker. Jika diagnosis tidak ditegakkan pada waktunya dengan perkembangan gejala neurologis, serangan akut dikaitkan dengan: berisiko tinggi dari kematian. Kebanyakan pasien sembuh total, tetapi gejala neurologis dapat bertahan selama berbulan-bulan atau bertahun-tahun. Yang paling penting adalah pencegahan serangan akut dengan menginstruksikan pasien untuk menghindari paparan faktor pencetus seperti obat-obatan, steroid, konsumsi alkohol, atau puasa yang disengaja.
T.P. Harison. prinsip penyakit dalam.Terjemahan d.m.s. A.V. Suchkova, Ph.D. N.N. Zavadenko, Ph.D. D.G. Katkovsky
Penyakit yang berhubungan dengan gangguan sintesis heme dan sering dimanifestasikan oleh anemia, sensitisasi kulit dan berbagai gangguan neurologis. Salah satu kasus pertama dijelaskan oleh Schultz pada abad ke-19 - 1874.
Berbagai jenis diidentifikasi porfiria, yang masing-masing berhubungan dengan defek pada salah satu dari delapan enzim yang terlibat dalam sintesis heme (kecuali 5-aminolevulinate synthetase). Gen yang mengkode enzim ini dan lokalisasi kromosomnya telah ditentukan. Sebagian besar kerusakan molekuler yang mendasari berbagai jenis penyakit telah diketahui.
Diagram biosintesis hemeALA - asam 5-aminolevulinat, PBG - porfobilinogen, UPG - uroporfirinogen, CPG - koproporfirinogen, PPG - protoporfirinogen
Blok biosintetik, akibat cacat enzimatik, paling menonjol di hati dan sumsum tulang - organ di mana jumlah utama heme disintesis. Setiap jenis porfiria dicirikan oleh gambaran klinis dan patologis yang mencerminkan defek pada enzim tertentu dan jenis pewarisannya.
Secara umum, untuk porfiria dua sindrom klinis utama adalah karakteristik: fotosensitifitas kulit dan sindrom gangguan neurologis. Fotosensitisasi kulit adalah hasil reaksi porfirin yang disimpan di kulit terhadap radiasi matahari. Gangguan neurologis disebabkan oleh peningkatan produksi dan ekskresi prekursor porfirin ALA dan porfobilinogen. Dengan cacat pada dua atau lebih enzim yang terlibat dalam sintesis heme, porfiria ganda didiagnosis.
Kelainan genetik dan metabolisme pada porfiria Catatan. 1) * - persentase nilai aktivitas biasa enzim; 2) metabolit utama dan rute ekskresi dicetak tebal; 3) singkatan: ALA - asam 5-aminolevulinat, PBG - porfobilinogen, UPG - uroporfirinogen, CPG - koproporfirinogen, PPG - protoporfirinogen.
Catatan. 1) * - persentase nilai aktivitas biasa enzim; 2) metabolit utama dan rute ekskresi dicetak tebal; 3) singkatan: ALA - asam 5-aminolevulinat, PBG - porfobilinogen, UPG - uroporfirinogen, CPG - koproporfirinogen, PPG - protoporfirinogen.
Klasifikasi Porfiria
SAYA. Porfiria dengan fotosensitifitas kulit:
- Porfiria eritropoietik kongenital
- Porfiria kulit lanjut
- Protoporfiria
II. Porfiria akut atau terinduksi:
- Porfiria dengan manifestasi neurologis
- Porfiria intermiten akut
- Porfiria ALC-D
- Porfiria dengan manifestasi neurologis dan kulit
- Porfiria beraneka ragam
- Koproporfiria
AKU AKU AKU. Porfiria ganda
Heme adalah kelompok prostetik dari banyak protein: hemoglobin, mioglobin, sitokrom CPE mitokondria, sitokrom P 450 yang terlibat dalam oksidasi mikrosomal. Enzim katalase, peroksidase, sitokrom oksidase mengandung heme sebagai koenzim.
Semua sel tubuh memiliki protein yang mengandung heme, oleh karena itu, sintesis heme terjadi di semua sel, kecuali eritrosit, yang, seperti diketahui, tidak memiliki sistem sintesis protein.
Pemecahan heme dalam sel RES menghasilkan pigmen empedu bilirubin. Katabolisme bilirubin lebih lanjut di hati, usus dan ginjal mengarah pada pembentukan produk akhir pemecahan heme stercobilin dan urobilin, yang masing-masing terkandung dalam feses dan urin. Besi yang dilepaskan selama pemecahan heme digunakan lagi untuk sintesis protein yang mengandung besi.
I. STRUKTUR DAN BIOSINTESIS HEMA a. struktur heme
Heme terdiri dari ion besi dan porfirin (Gbr. 13-1). Struktur porfirin didasarkan pada porfin. Porfin terdiri dari empat cincin pirol yang dihubungkan oleh jembatan methen (Gbr. 13-1). Tergantung pada struktur substituen dalam cincin pirol, beberapa jenis porfirin dibedakan: protoporfirin, etioporfirin, mesoporfirin, dan koproporfirin. Protoporfirin adalah prekursor dari semua jenis porfirin lainnya.
Heme dari berbagai protein mungkin mengandung: jenis yang berbeda porfirin (lihat bagian 6). Heme hemoglobin mengandung protoporfirin IX, yang memiliki 4 metil, 2 radikal vinil dan 2 residu asam propionat. Besi dalam heme berada dalam keadaan tereduksi (Fe +2) dan terikat oleh dua ikatan kovalen dan dua ikatan koordinasi dengan atom nitrogen dari cincin pirol. Ketika besi dioksidasi, heme diubah
menjadi hematin (Fe3+). Jumlah heme terbesar terdapat pada eritrosit yang berisi hemoglobin, sel otot dengan mioglobin, dan sel hati karena tingginya kandungan sitokrom P 450 di dalamnya.
B. biosintesis heme
Heme disintesis di semua jaringan, tetapi pada tingkat tertinggi di sumsum tulang dan hati (Gbr. 13-2). Di sumsum tulang, heme diperlukan untuk sintesis hemoglobin dalam retikulosit, dalam hepatosit - untuk pembentukan sitokrom P 450.
Reaksi sintesis heme pertama - pembentukan asam 5-aminolevulinat dari glisin dan suksinil-KoA (Gbr. 13-3) berlangsung di matriks mitokondria, di mana salah satu substrat dari reaksi ini, suksinil-KoA, dibentuk di CTC. Reaksi ini dikatalisis oleh enzim 5-aminolevulinate sintase yang bergantung pada piridoksal.
Dari mitokondria, asam 5-aminolevulinat memasuki sitoplasma. Di sitoplasma, tahap menengah sintesis heme terjadi: kombinasi 2 molekul asam 5-aminolevulinat menjadi molekul porfobilinogen (Gbr. 13-4), deaminasi porfobilinogen dengan pembentukan hidroksimetilbilana, konversi enzimatik hidroksimetilbilana menjadi molekul dari uroporphobilinogen III, dekarboksilasi yang terakhir dengan pembentukan coproporphyrinogen III. Hidroksimetilbilana juga dapat secara non-enzimatis berubah menjadi uroporfirinogen I, yang didekarboksilasi menjadi koproporfirinogen I. Dari sitoplasma, koproporfirinogen III kembali memasuki mitokondria, tempat berlangsungnya reaksi sintesis heme terakhir. Sebagai hasil dari dua berturut-turut reaksi oksidatif co-roporfirinogen III diubah menjadi protoporfirinogen IX, dan protoporfirinogen IX diubah menjadi protoporfirin IX. Enzim ferrochelatase, yang mengikat besi divalen ke protoporfirin IX, mengubahnya menjadi heme (Gbr. 13-2). Sumber zat besi untuk sintesis heme adalah besi-depositing protein feritin. Sin-
Beras. 13-1. Struktur porphin (A), protoporphyrin IX (B), dan hemoglobin heme (C). Porfin adalah struktur siklik yang terdiri dari empat cincin pirol yang dihubungkan oleh jembatan methen. Protoporfirin IX memiliki empat metil, dua radikal vinil dan dua residu asam propionat. Dalam heme hemoglobin, Fe 2+ membentuk dua ikatan kovalen dan dua koordinasi dengan atom nitrogen dari cincin pirol protoporfirin IX.
heme yang disintesis bergabung dengan rantai - dan -po-lipepeptide globin untuk membentuk hemoglobin. Heme mengatur sintesis globin: dengan penurunan laju sintesis heme, sintesis globin dalam retikulosit terhambat.
B. REGULASI HEME BIOSINTESIS
Reaksi regulasi sintesis heme dikatalisis oleh enzim 5-aminolevulinate sintase yang bergantung pada piridoksal. Laju reaksi diatur secara alosterik dan pada tingkat translasi enzim.
Heme adalah penghambat alosterik dan korepresor sintesis 5-aminolevulinat sintase (Gbr. 13-5).
Dalam retikulosit, sintesis enzim ini pada tahap translasi diatur oleh zat besi. Di tempat inisiasi mRNA yang mengkode enzim, terdapat urutan nukleotida yang membentuk lingkaran jepit rambut, yang disebut elemen peka-besi (dari bahasa Inggris. elemen responsif besi, IRE) (Gbr. 13-6).
Pada konsentrasi tinggi besi dalam sel, ia membentuk kompleks dengan residu sistein dari protein pengikat besi pengatur. Interaksi besi dengan protein pengikat besi pengatur menyebabkan penurunan afinitas protein ini untuk elemen IRE dari mRNA yang mengkode 5-aminolevulinate sintase dan kelanjutan translasi (Gbr. 13-6, A). Pada konsentrasi besi yang rendah, pengikatan besi

Beras. 13-2. Sintesis hem. Angka-angka dalam diagram menunjukkan enzim: 1 - 5-aminolevulinate sintase; 2 - 5-aminole-vulinate dehidratase; 3 - deaminase porfobilinogen; 4 - uroporfirinogen III cosynthase; 5 - dekarboksilase gen uroporfirin; 6 - koproporfirinogen III oksidase; 7 - protoporfirinogen oksidase; 8 - ferrochelatase. Huruf menunjukkan substituen dalam cincin pirol: M - metil, B - vinil, P - residu asam propionat, A - asetil, PF - piridoksal fosfat. Feritin, protein yang menyimpan zat besi dalam sel, berfungsi sebagai donor zat besi.

Beras. 13-3. Reaksi pembentukan asam 5-aminolevulinat.
protein menempel pada elemen peka-besi yang terletak di ujung mRNA yang tidak diterjemahkan 5', dan translasi sintase 5-aminolevulinat dihambat (Gbr. 13-6, B).
5-Aminolevulinate dehidratase juga dihambat secara alosterik oleh heme, tetapi karena aktivitas enzim ini hampir 80 kali lebih tinggi daripada aktivitas 5-aminolevulinate sintase, ini adalah sedikit signifikansi fisiologis.

Beras. 13-4. Reaksi pembentukan porfobilinogen.

Beras. 13-5. Regulasi sintesis heme dan hemoglobin. Heme, dengan prinsip umpan balik negatif, menghambat 5-aminolevulinate sintase dan 5-aminolevulinate dehidratase dan merupakan penginduksi translasi α- Dan β rantai hemoglobin.

Beras. 13-6. Regulasi sintesis aminolevulinate sintase. A - at konsentrasi tinggi besi dalam retikulosit, ia menempel pada protein pengikat besi dan mengurangi afinitas protein ini untuk RNA utusan elemen peka-besi (IRE) yang mengkode 5-aminolevulinate sintase. Faktor inisiasi translasi protein mengikat mRNA dan memulai translasi sintase 5-aminolevulinate. B - dengan kandungan besi yang rendah dalam retikulosit, protein pengikat besi memiliki afinitas tinggi untuk IRE dan berinteraksi dengannya. Faktor inisiasi translasi protein tidak dapat menempel pada mRNA, dan translasi berhenti.
Defisiensi piridoksal fosfat dan obat-obatan, yang merupakan analog strukturalnya, mengurangi aktivitas sintase 5-amino-levulinat.
D. GANGGUAN BIOSINTESIS HEMA. PORFIRIA
Gangguan sintesis heme herediter dan didapat, disertai dengan peningkatan kandungan porfirinogen, serta produk oksidasi mereka dalam jaringan dan darah dan penampilannya dalam urin, disebut porfiria ("porfirin" dalam bahasa Yunani berarti ungu). Urine pasien berwarna merah.
Porfiria herediter disebabkan oleh cacat genetik pada enzim yang terlibat
dalam sintesis heme, dengan pengecualian 5-aminolevulinate sintase. Pada penyakit ini, penurunan pembentukan heme dicatat. Karena heme adalah penghambat alosterik sintase 5-aminolevulinat, aktivitas enzim ini meningkat, dan ini mengarah pada akumulasi produk antara sintesis heme - asam 5-aminolevulinat dan porfirinogen.
Tergantung pada lokasi utama proses patologis membedakan antara porfiria herediter hepatik dan eritropoietik. Porfiria eritropoietik disertai dengan akumulasi porfirin dalam normoblas dan eritrosit, dan porfiria hati - dalam hepatosit.
Dalam bentuk porfiria yang parah, gangguan neuropsikis, disfungsi RES, dan kerusakan kulit diamati. Porfirinogen tidak berwarna dan tidak berfluoresensi, tetapi dalam cahaya mereka mudah diubah menjadi porfirin. Yang terakhir menunjukkan fluoresensi merah intens di bawah sinar ultraviolet. Di kulit di bawah sinar matahari, sebagai hasil interaksi dengan porfirin, oksigen masuk ke keadaan singlet. Oksigen singlet menyebabkan percepatan peroksidasi lipid membran sel dan penghancuran sel; oleh karena itu, porfiria sering disertai dengan fotosensitifitas dan ulserasi pada area kulit yang terbuka. Gangguan neuropsikiatri pada porfiria dikaitkan dengan fakta bahwa 5-aminolevulinat dan porfirinogen adalah neurotoksin.
Kadang-kadang, dalam bentuk ringan porfiria herediter, penyakit ini mungkin tidak menunjukkan gejala, tetapi penggunaan obat yang menginduksi sintesis 5-aminolevulinate synthase dapat menyebabkan eksaserbasi penyakit. Penginduksi sintesis 5-aminolevulinate synthase adalah seperti: obat-obatan yang dikenal, seperti sulfonamid, barbiturat, diklofenak, voltaren, steroid, gestagens. Dalam beberapa kasus, gejala penyakit tidak muncul sampai pubertas, ketika peningkatan pembentukan -steroid menyebabkan induksi sintesis 5-aminolevulinate synthase. Porfiria juga diamati dalam kasus keracunan dengan garam timbal, karena timbal menghambat 5-aminolevulinate dehidratase dan ferrochelatase. Beberapa herbisida dan insektisida yang mengandung halogen merupakan penginduksi sintesis 5-aminolevulinate sintase, sehingga konsumsinya disertai dengan gejala porfiria.
II. PERTUKARAN BESI
Tubuh orang dewasa mengandung 3-4 g zat besi, yang hanya sekitar 3,5 mg
ada di plasma darah. Hemoglobin memiliki sekitar 68% zat besi dari seluruh tubuh, feritin - 27%, mioglobin - 4%, transferin - 0,1%, Semua enzim yang mengandung zat besi hanya menyumbang 0,6% dari zat besi yang ada dalam tubuh. Sumber zat besi dalam biosintesis protein yang mengandung zat besi adalah zat besi makanan dan zat besi yang dilepaskan selama pemecahan eritrosit secara konstan di sel-sel hati dan limpa.
Dalam lingkungan netral atau basa, besi berada dalam keadaan teroksidasi - Fe 3+ , membentuk kompleks besar yang mudah beragregasi dengan OH - , anion lain dan air. Pada nilai pH rendah, besi tereduksi dan mudah terdisosiasi. Proses reduksi dan oksidasi besi memastikan redistribusi antara makromolekul dalam tubuh. Ion besi memiliki afinitas tinggi terhadap banyak senyawa dan membentuk kompleks kelat dengannya, mengubah sifat dan fungsi senyawa ini, sehingga pengangkutan dan pengendapan besi dalam tubuh dilakukan oleh protein khusus. Dalam sel, besi disimpan oleh protein feritin; dalam darah, diangkut oleh protein transferin.
tetapi. penyerapan zat besi di usus
Dalam makanan, besi terutama dalam keadaan teroksidasi (Fe 3+) dan merupakan bagian dari protein atau garam. asam organik. Pelepasan besi dari garam asam organik difasilitasi oleh lingkungan asam jus lambung. Jumlah terbesar besi diserap di duodenum. Asam askorbat yang terkandung dalam makanan mengembalikan zat besi dan meningkatkan penyerapannya, karena hanya Fe 2+ yang memasuki sel-sel mukosa usus. Jumlah makanan harian biasanya mengandung 15-20 mg zat besi, dan hanya sekitar 10% dari jumlah ini yang diserap. Tubuh orang dewasa kehilangan sekitar 1 mg zat besi per hari.
Jumlah zat besi yang diserap ke dalam sel-sel mukosa usus, sebagai suatu peraturan, melebihi kebutuhan tubuh. Aliran besi dari enterosit ke dalam darah tergantung pada kecepatan sintesis protein apoferritin di dalamnya. Apoferritin "menjebak" besi dalam enterosit dan berubah menjadi feritin, yang tetap berada di enterosit. Dengan cara ini, alirannya berkurang
pelepasan besi ke dalam kapiler darah dari sel-sel usus. Ketika kebutuhan zat besi rendah, laju sintesis apoferritin meningkat (lihat di bawah "Pengaturan masuknya zat besi ke dalam sel"). Pelepasan konstan sel mukosa ke dalam lumen usus membebaskan tubuh dari kelebihan zat besi. Dengan kekurangan zat besi dalam tubuh, apoferritin hampir tidak disintesis dalam enterosit. Besi yang berasal dari enterosit ke dalam darah mengangkut protein plasma darah transferin (Gbr. 13-7).
B. TRANSPORTASI BESI DALAM PLASMA DARAH DAN MASUKNYA KE SEL
Dalam plasma, besi mengangkut protein transferin. Transferin adalah glikoprotein yang disintesis di hati dan hanya mengikat besi teroksidasi (Fe 3+). Besi yang memasuki aliran darah dioksidasi oleh enzim ferroxidase, yang dikenal sebagai protein plasma yang mengandung tembaga seruloplasmin. Satu molekul transferin dapat mengikat satu atau dua ion Fe 3+, tetapi secara bersamaan dengan anion CO 3 2- untuk membentuk kompleks transferrin-2 (Fe 3+ -CO 3 2-). Biasanya, transferin darah jenuh dengan besi sekitar 33%.
Transferin berinteraksi dengan reseptor membran sel spesifik.
Sebagai hasil dari interaksi ini, kompleks Ca2+ -calmodulin-PKC terbentuk di sitosol sel, yang memfosforilasi reseptor transferin dan menyebabkan pembentukan endosom. Pompa proton yang bergantung pada ATP yang terletak di membran endosom menciptakan lingkungan asam di dalam endosom. Dalam lingkungan asam endosom, besi dilepaskan dari transferin. Setelah itu, kompleks reseptor-apotransferrin kembali ke permukaan membran plasma sel. Pada nilai pH netral cairan ekstraseluler, apotransferin mengubah konformasinya, memisahkan diri dari reseptor, memasuki plasma darah dan menjadi mampu mengikat ion besi lagi dan dimasukkan dalam siklus baru pengangkutannya ke dalam sel. Besi dalam sel digunakan untuk sintesis protein yang mengandung besi atau disimpan dalam protein feritin.
Feritin adalah protein oligomer dengan berat molekul 500 kD. Ini terdiri dari rantai polipeptida berat (21 kD) dan ringan (19 kD) yang membentuk 24 protomer. Seperangkat protomer yang berbeda dalam oligomer feritin menentukan pembentukan beberapa isoform protein ini di jaringan yang berbeda. Feritin adalah bola berongga yang dapat berisi hingga 4500 ion besi, tetapi biasanya mengandung kurang dari 3000. Rantai berat

Beras. 13-7. Masuknya besi eksogen ke dalam jaringan. Di rongga usus, zat besi dilepaskan dari protein dan garam asam organik dalam makanan. Penyerapan zat besi dipromosikan oleh asam askorbat, yang mengembalikan zat besi. Di dalam sel mukosa usus, kelebihan besi yang masuk bergabung dengan protein apoferritin membentuk feritin, sedangkan feritin mengoksidasi Fe2+ menjadi Fe3+. Masuknya besi dari sel-sel mukosa usus ke dalam darah disertai dengan oksidasi besi oleh enzim ferrooksidase dalam serum darah. Dalam darah, Fe3+ mengangkut transferin protein serum. Dalam jaringan, Fe2+ digunakan untuk sintesis protein yang mengandung besi atau disimpan dalam feritin.
feritin mengoksidasi Fe 2+ menjadi Fe 3+ . Besi dalam bentuk hidroksida fosfat terletak di tengah bola, yang cangkangnya dibentuk oleh bagian protein molekul. Ia masuk dan dikeluarkan melalui saluran yang menembus cangkang protein apoferritin, tetapi besi juga dapat disimpan di bagian protein dari molekul feritin. Feritin ditemukan di hampir semua jaringan, tetapi dalam jumlah terbesar di hati, limpa dan sumsum tulang. Bagian yang tidak signifikan dari feritin diekskresikan dari jaringan ke dalam plasma darah. Karena aliran feritin ke dalam darah sebanding dengan kandungannya dalam jaringan, konsentrasi feritin dalam darah merupakan indikator diagnostik penting dari simpanan besi dalam tubuh pada anemia defisiensi besi. Metabolisme besi dalam tubuh ditunjukkan pada Gambar. 13-8.
B. PENGATURAN PENGHASILAN BESI KE SEL
Kandungan besi dalam sel ditentukan oleh rasio laju masuk, penggunaan, dan deposisi dan dikendalikan oleh dua mekanisme molekuler. Kecepatan masuknya besi ke dalam sel non-eritroid bergantung pada jumlah protein reseptor transferin dalam membrannya. Kelebihan zat besi dalam sel menyimpan feritin. Sintesis reseptor apoferritin dan transferin diatur pada tingkat translasi protein ini dan bergantung pada kandungan besi di dalam sel.
Pada ujung 3' mRNA reseptor transferin yang tidak diterjemahkan dan pada ujung 5' mRNA apoferritin yang tidak diterjemahkan, terdapat loop jepit rambut - elemen peka besi IRE (Gbr. 13-9 dan 13-10). Selain itu, mRNA dari reseptor transfer

Beras. 13-8. Metabolisme zat besi dalam tubuh.

Beras. 13-9. Regulasi sintesis apoferritin. A - dengan penurunan kandungan besi dalam sel, protein pengikat besi memiliki afinitas tinggi untuk IRE dan berinteraksi dengannya. Ini mencegah perlekatan faktor protein inisiasi translasi ke mRNA yang mengkode apoferritin, dan sintesis apoferritin berhenti; B - dengan peningkatan kandungan zat besi dalam sel, ia berinteraksi dengan protein pengikat besi, akibatnya afinitas protein ini untuk IRE berkurang. Faktor inisiasi translasi protein menempel pada mRNA yang mengkode apoferritin dan memulai translasi apoferritin.
rina memiliki 5 loop, sedangkan apoferritin mRNA hanya memiliki 1.
Daerah mRNA ini dapat berinteraksi dengan protein pengikat IRE pengatur. Pada konsentrasi besi yang rendah di dalam sel, protein pengikat IRE berikatan dengan apoferritin mRNA IRE dan mencegah perlekatan faktor protein inisiasi translasi (Gbr. 13-9, A). Akibatnya, kecepatan translasi apoferritin dan kandungannya di dalam sel menurun. Pada saat yang sama, pada konsentrasi besi yang rendah di dalam sel, protein pengikat IRE mengikat elemen sensitif besi dari mRNA reseptor transferin dan mencegah penghancurannya oleh enzim RNase (Gbr. 13-10, A). Hal ini menyebabkan peningkatan jumlah reseptor
transferin dan percepatan masuknya besi ke dalam sel.
Dengan peningkatan kandungan zat besi dalam sel sebagai akibat interaksinya dengan protein pengikat IRE, gugus SH dari pusat aktif protein ini dioksidasi dan afinitas terhadap elemen mRNA yang peka terhadap zat besi berkurang. Ini mengarah pada dua konsekuensi:
Pertama, translasi apoferritin dipercepat (Gbr. 13-9, B);
Kedua, protein pengikat IRE melepaskan loop jepit rambut dari mRNA reseptor transferin, dan protein tersebut dihancurkan oleh enzim RNase, sebagai akibatnya, kecepatan sintesis reseptor transferin menurun (Gbr. 1310, B). Percepatan sintesis apoferritin dan penghambatan sintesis trans-

Beras. 13-10. Regulasi sintesis reseptor transferin. A - pada kandungan besi yang rendah di dalam sel, protein sensitif besi memiliki afinitas tinggi untuk mRNA IRE yang mengkode protein reseptor transferin. Perlekatan protein pengikat besi ke mRNA IRE mencegah penghancurannya oleh RNase dan sintesis protein reseptor transferrin berlanjut; B - At konten tinggi besi di dalam sel, afinitas protein pengikat besi untuk IRE menurun, dan mRNA menjadi tersedia untuk aksi RNase, yang menghidrolisisnya. Penghancuran mRNA menyebabkan penurunan sintesis protein reseptor transferin.
ferrin menyebabkan penurunan konten
besi di dalam sel. Secara umum, mekanisme ini mengatur kandungan besi dalam sel dan penggunaannya untuk sintesis protein yang mengandung besi.
D. GANGGUAN METABOLISME BESI
Anemia defisiensi besi dapat terjadi dengan perdarahan berulang, kehamilan, sering melahirkan, borok, dan tumor.
Saluran pencernaan, setelah operasi pada saluran pencernaan. Dengan anemia defisiensi besi, ukuran eritrosit dan pigmentasinya berkurang (eritrosit hipokromik berukuran kecil). Dalam eritrosit, kandungan hemoglobin menurun, saturasi transferin dengan besi menurun, dan konsentrasi feritin dalam jaringan menurun. Alasan untuk perubahan ini adalah kekurangan zat besi dalam tubuh, akibatnya sintesis heme dan feritin di jaringan non-eritroid dan hemoglobin dalam sel eritroid menurun.
Hemokromatosis. Ketika jumlah besi dalam sel melebihi volume depot feritin, besi disimpan di bagian protein dari molekul feritin. Sebagai hasil dari pembentukan endapan amorf kelebihan zat besi, feritin diubah menjadi hemosiderin. Hemo-siderin kurang larut dalam air dan mengandung hingga 37% zat besi. Akumulasi butiran hemosiderin di hati, pankreas, limpa menyebabkan kerusakan pada organ-organ ini - hemokromatosis. Hemokromatosis mungkin karena peningkatan herediter dalam penyerapan zat besi di usus, sedangkan kandungan zat besi dalam tubuh pasien bisa mencapai 100 g. Penyakit ini diturunkan secara resesif autosomal, dan sekitar 0,5% bule homozigot untuk hemokromatosis. gen. Akumulasi hemosiderin di pankreas menyebabkan penghancuran sel pulau Langerhans dan, sebagai akibatnya, menjadi diabetes mellitus. Deposisi hemosiderin dalam hepatosit menyebabkan sirosis hati, dan pada miokardiosit - gagal jantung. Pasien dengan hemochromatosis herediter dirawat dengan pertumpahan darah secara teratur, mingguan atau sebulan sekali, tergantung pada tingkat keparahan kondisi pasien. Transfusi darah yang sering dapat menyebabkan hemochromatosis, dalam kasus ini, pasien dirawat dengan obat pengikat besi.
AKU AKU AKU. KATABOLISME HEMOGLOBIN
Eritrosit memiliki umur yang pendek (sekitar 120 hari). Dalam kondisi fisiologis, sekitar 1-2x10 eritrosit per hari dihancurkan dalam tubuh orang dewasa. Katabolismenya terjadi terutama di sel retikuloendotelial limpa, kelenjar getah bening, sumsum tulang dan hati. Dengan penuaan eritrosit, kandungan asam sialat dalam komposisi glikoprotein membran plasma berkurang. Komponen karbohidrat yang berubah dari glikoprotein membran eritrosit diikat oleh reseptor sel RES, dan eritrosit "dibenamkan" di dalamnya oleh endositosis. Pemecahan eritrosit dalam sel-sel ini dimulai dengan pemecahan hemoglobin menjadi heme dan globin dan selanjutnya hidrolisis bagian protein hemoglobin oleh enzim lisosom.
A. KATABOLISME HEMA
Reaksi pertama katabolisme heme terjadi dengan partisipasi kompleks enzimatik yang bergantung pada NADPH oksigenase heme. Sistem enzim terlokalisasi di membran RE, di area rantai transpor elektron oksidasi mikrosomal. Enzim mengkatalisis pemutusan ikatan antara dua cincin pirol yang mengandung residu vinil, sehingga mengungkapkan struktur cincin (Gbr. 13-11). Selama reaksi, peran tetrapir linier terbentuk - biliverdin(pigmen kuning) dan karbon monoksida (CO), yang diperoleh dari karbon gugus metenil. Heme menginduksi transkripsi gen heme oksigenase, yang benar-benar spesifik untuk heme.
Ion besi yang dilepaskan selama pemecahan heme dapat digunakan untuk sintesis molekul hemoglobin baru atau untuk sintesis protein lain yang mengandung besi. Biliverdin direduksi menjadi bilirubin oleh enzim yang bergantung pada NADPH biliverdin reduktase. Bilirubin dibentuk tidak hanya dari pemecahan hemoglobin, tetapi juga dari katabolisme protein lain yang mengandung heme, seperti sitokrom dan mioglobin. Dengan pemecahan 1 g hemoglobin, 35 mg bilirubin terbentuk, dan sekitar 250-350 mg bilirubin per hari pada orang dewasa. Metabolisme bilirubin lebih lanjut terjadi di hati.
B. METABOLISME BILIRUBIN
Bilirubin, yang dibentuk dalam sel-sel RES (limpa dan sumsum tulang), kurang larut dalam air, diangkut melalui darah dalam kombinasi dengan albumin protein plasma darah. Bentuk bilirubin ini disebut bilirubin tak terkonjugasi. Setiap molekul albumin mengikat 2 (atau bahkan 3) molekul bilirubin, salah satunya terikat lebih erat dengan protein (afinitas lebih tinggi) daripada yang lain. Dengan pergeseran pH darah ke sisi asam (peningkatan konsentrasi badan keton, laktat), muatan dan konformasi albumin berubah, dan afinitas terhadap bilirubin menurun. Oleh karena itu, bilirubin, yang terikat secara longgar pada albumin, dapat dipindahkan dari pusat pengikatan dan membentuk kompleks dengan kolagen matriks ekstraseluler dan lipid membran. Sejumlah senyawa obat bersaing dengan bilirubin untuk

Beras. 13-11. Kerusakan heme. M - (-CH 3) - gugus metil; B - (-CH \u003d CH 2) - grup vinil; P - (-CH 2 -CH 2 -COOH) - residu asam propionat. Selama reaksi, satu gugus metil diubah menjadi karbon monoksida dan dengan demikian struktur cincin terungkap. Biliverdin yang terbentuk di bawah aksi biliverdin reduktase diubah menjadi bilirubin.
pusat albumin berafinitas tinggi dan berafinitas tinggi.
Absorbsi bilirubin oleh sel parenkim hati
Kompleks albumin-bilirubin, dikirim dengan aliran darah ke hati, di permukaan
membran plasma hepatosit terdisosiasi. Bilirubin yang dilepaskan membentuk kompleks sementara dengan lipid membran plasma. Difusi terfasilitasi dari bilirubin ke dalam hepatosit dilakukan oleh dua jenis protein pembawa: ligandin (mengangkut jumlah utama empedu-
ruby) dan protein Z. Aktivitas pengambilan bilirubin oleh hepatosit bergantung pada kecepatan metabolismenya di dalam sel.
Ligandin dan protein Z juga ditemukan di sel-sel ginjal dan usus, oleh karena itu, dalam kasus insufisiensi fungsi hati, mereka mampu mengkompensasi melemahnya proses detoksifikasi di organ ini.
Konjugasi bilirubin di RE halus
Dalam RE halus hepatosit, residu asam glukuronat melekat pada bilirubin - suatu reaksi konjugasi. Bilirubin memiliki 2 gugus karboksil, oleh karena itu, dapat bergabung dengan 2 molekul asam glukuronat, membentuk konjugat yang sangat larut dalam air - bilirubin di-glukuronida (bilirubin terkonjugasi, atau langsung) (Gbr. 13-12).
Donor asam glukuronat adalah UDP-glukuronat. Enzim spesifik, UDP-glucuronyltransferases (uridin diphosphoglucuronyltransferases) mengkatalisis pembentukan bilirubin mono- dan diglucuronides (Gbr. 13-13). Beberapa obat, seperti fenobarbital, berfungsi sebagai penginduksi untuk sintesis UDP-glucuronyltransferases (lihat bagian 12).
Sekresi bilirubin ke dalam empedu
Sekresi bilirubin terkonjugasi ke dalam empedu mengikuti mekanisme transpor aktif, yaitu melawan gradien konsentrasi. Transpor aktif mungkin merupakan langkah pembatas kecepatan dalam keseluruhan proses metabolisme bilirubin di hati. Diglucuronide normal

Beras. 13-12. Struktur bilirubindiglucuronide (bilirubin "langsung" terkonjugasi). Asam glukuronat adalah eter yang terikat pada dua residu asam propionat untuk membentuk asilglukuronida.
bilirubin adalah bentuk utama ekskresi bilirubin ke dalam empedu, tetapi keberadaan sejumlah kecil mono-glukuronida tidak dikecualikan. Pengangkutan bilirubin terkonjugasi dari hati ke empedu diaktifkan oleh obat yang sama yang dapat menginduksi konjugasi bilirubin. Dengan demikian, dapat dikatakan bahwa laju konjugasi bilirubin dan transpor aktif bilirubin glukuronida dari hepatosit ke empedu saling berhubungan erat (Gbr. 13-14).
B. KATABOLISME BILIRUBINDIGLUCURONIDE
Di usus, bilirubin glukuronida yang masuk dihidrolisis oleh enzim bakteri spesifik -glukuronidase, yang menghidrolisis ikatan antara bilirubin dan residu asam glukuronat. Gratis-

Beras. 13-13. Pembentukan bilirubin diglukuronida.

Beras. 13-14. Siklus bilirubin-urobilinogen di hati. 1 - Katabolisme Hb di sel retikuloendotelial sumsum tulang, limpa, kelenjar getah bening; 2 - pembentukan bentuk transportasi kompleks bilirubin-albumin; 3 - masuknya bilirubin ke dalam hati; 4 - pembentukan bilirubinglucuronides;
5 - sekresi bilirubin sebagai bagian dari empedu ke dalam usus;
6 - katabolisme bilirubin di bawah aksi bakteri usus; 7 - penghapusan urobilinogen dengan tinja; 8 - penyerapan urobilinogen ke dalam darah; 9 - asimilasi urobilinogen oleh hati; 10 - masuknya sebagian urobilinogen ke dalam darah dan ekskresi oleh ginjal dengan urin; 11 - sebagian kecil urobilinogen disekresikan ke dalam empedu.
bilirubin, yang diproduksi selama reaksi ini, dipulihkan di bawah aksi mikroflora usus dengan pembentukan sekelompok senyawa tetrapirol tidak berwarna - urobilinogen(Gbr. 13-15).
Di ileum dan usus besar, sebagian kecil urobilinogen diserap kembali, memasuki hati dengan darah vena portal. Bagian utama urobilinogen dari hati dalam komposisi empedu diekskresikan ke dalam usus dan diekskresikan dengan
kotoran dari tubuh, bagian dari urobilinogen dari hati memasuki aliran darah dan dikeluarkan dalam urin dalam bentuk urobilin (Gbr. 13-14). Biasanya, sebagian besar urobilinogen tidak berwarna yang diproduksi di usus besar dioksidasi di rektum menjadi pigmen coklat di bawah aksi mikroflora usus. urobilin dan dibuang bersama feses. Warna feses disebabkan oleh adanya urobilin.

Beras. 13-15. Struktur beberapa pigmen empedu. Mesobilinogen adalah produk antara katabolisme bilirubin di usus.
iv. nilai diagnostik
menentukan konsentrasi bilirubin dalam cairan biologis manusia
Saat ini, untuk menentukan kandungan bilirubin dalam serum (plasma) darah, metode yang diusulkan pada tahun 1916 oleh Van der Berg untuk penentuan bilirubin dalam serum darah, berdasarkan diazoreaksi, digunakan.
Dalam keadaan normal, konsentrasi bilirubin total dalam plasma adalah 0,3-1 mg/dL (1,7-17 mol/L), 75% dari total Bilirubin dalam bentuk tidak terkonjugasi (bilirubin tidak langsung). Di klinik, bilirubin terkonjugasi disebut langsung karena larut dalam air dan dapat dengan cepat berinteraksi dengan reagen diazo, membentuk senyawa merah muda - ini adalah reaksi Van der Berg langsung. Bilirubin tak terkonjugasi bersifat hidrofobik, oleh karena itu terkandung dalam plasma darah dalam kompleks dengan albumin dan tidak bereaksi dengan reagen diazo sampai pelarut organik, seperti etanol, ditambahkan, yang mengendapkan albumin. Bilirubin tak terkonjugasi yang bereaksi dengan pewarna azo hanya setelah pengendapan protein disebut bilirubin tidak langsung.
Pada pasien dengan patologi hepatoseluler, disertai dengan peningkatan yang berkepanjangan
konsentrasi bilirubin terkonjugasi dalam darah, bentuk ketiga dari bilirubin plasma ditemukan, di mana bilirubin terikat secara kovalen dengan albumin, dan karena itu tidak dapat dipisahkan dengan cara biasa. Dalam beberapa kasus, hingga 90% dari total bilirubin darah mungkin dalam bentuk ini.
A. penyakit kuning
Penyebab hiperbilirubinemia dapat berupa peningkatan produksi bilirubin di luar kemampuan hati untuk mengekskresikannya, atau kerusakan hati yang menyebabkan gangguan sekresi bilirubin ke dalam empedu dalam jumlah normal. Hiperbilirubinemia juga dicatat dengan penyumbatan saluran empedu hati.
Dalam semua kasus, kandungan bilirubin total dalam darah meningkat. Ketika konsentrasi tertentu tercapai, ia berdifusi ke dalam jaringan, menodainya dengan warna kuning. Menguningnya jaringan karena pengendapan bilirubin di dalamnya disebut penyakit kuning. Secara klinis, penyakit kuning mungkin tidak muncul sampai konsentrasi bilirubin dalam plasma darah melebihi batas atas norma lebih dari 2,5 kali, yaitu. tidak akan melebihi 50 mol/l.
1. Ikterus hemolitik (prehepatik)
Diketahui bahwa kemampuan hati untuk membentuk glukuronida dan melepaskannya ke dalam empedu adalah 3-4 kali lebih tinggi daripada pembentukannya dalam kondisi fisiologis. Ikterus hemolitik (prehepatik) adalah hasil dari hemolisis sel darah merah yang intens. Hal ini disebabkan oleh pembentukan bilirubin yang berlebihan, melebihi
kemampuan hati untuk mengeluarkannya. Ikterus hemolitik berkembang ketika kapasitas cadangan hati habis. Penyebab utama ikterus suprahepatik adalah herediter atau didapat anemia hemolitik. Dengan anemia hemolitik yang disebabkan oleh sepsis, penyakit radiasi, defisiensi glukosa-6-fosfat dehidrogenase eritrosit, talasemia, transfusi golongan darah yang tidak kompatibel, keracunan sulfonamida, jumlah hemoglobin yang dilepaskan dari eritrosit per hari dapat mencapai hingga 45 g
(pada tingkat 6,25 g), yang secara signifikan meningkatkan pembentukan bilirubin. Hiperbilirubinemia pada pasien dengan ikterus hemolitik disebabkan oleh peningkatan yang signifikan (103-171 mol/l) dalam konsentrasi darah dari bilirubin tak terkonjugasi yang terikat albumin (bilirubin tidak langsung). Pembentukan di hati dan masuknya bilirubinglucuronides dalam jumlah besar ke usus (bilirubin langsung) menyebabkan peningkatan pembentukan dan ekskresi urobilinogen dengan feses dan urin dan warnanya yang lebih intens (Gbr. 13-16).
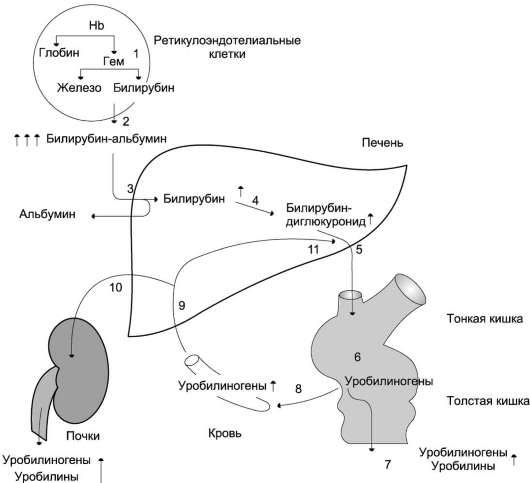
Beras. 13-16. Siklus bilirubin-urobilinogen pada ikterus hemolitik. 1 - Katabolisme Hb berlangsung dengan kecepatan yang meningkat; 2 - dalam darah, konsentrasi tidak bilirubin langsung; 3 - albumin dilepaskan dari kompleks bilirubin-albumin; 4 - aktivitas reaksi glukuronidasi meningkat, tetapi lebih rendah dari laju pembentukan bilirubin; 5 - sekresi bilirubin ke dalam empedu meningkat; 6, 7,10 - peningkatan kandungan urobilinogen dalam tinja dan urin memberi mereka warna yang lebih intens; urobilinogen diserap dari usus ke dalam darah (8) dan masuk ke hati melalui vena portal (9).
Salah satu tanda utama ikterus hemolitik adalah peningkatan kandungan bilirubin tak terkonjugasi (tidak langsung) dalam darah. Hal ini memudahkan untuk membedakannya dari ikterus mekanis (subhepatik) dan hepatoseluler (hepatik).
Bilirubin tak terkonjugasi bersifat toksik. Bilirubin tak terkonjugasi hidrofobik, lipofilik, mudah larut dalam lipid membran dan akibatnya menembus ke dalam mitokondria, memisahkan respirasi dan fosforilasi oksidatif di dalamnya, mengganggu sintesis protein, aliran ion kalium melalui membran sel dan organel. Ini secara negatif mempengaruhi keadaan sistem saraf pusat, menyebabkan sejumlah gejala neurologis yang khas pada pasien.
Penyakit kuning pada bayi baru lahir
Jenis ikterus hemolitik pribadi pada bayi baru lahir adalah "ikterus fisiologis", yang diamati pada hari-hari pertama kehidupan seorang anak. Alasan peningkatan konsentrasi Bilirubin tidak langsung dalam darah dipercepat hemolisis dan insufisiensi fungsi protein dan enzim hati yang bertanggung jawab untuk penyerapan, konjugasi dan sekresi bilirubin langsung. Pada bayi baru lahir, tidak hanya aktivitas UDP-glucuronyltransferase yang berkurang, tetapi, tampaknya, sintesis substrat kedua dari reaksi konjugasi UDP-glucuronate tidak cukup aktif.
UDP-glucuronyltransferase dikenal sebagai enzim yang dapat diinduksi (lihat Bagian 12). Bayi baru lahir dengan ikterus fisiologis diberikan obat fenobarbital, yang efek induksinya telah dijelaskan di bagian 12.
Salah satu komplikasi yang tidak menyenangkan penyakit kuning fisiologis» - ensefalopati bilirubin. Ketika konsentrasi bilirubin tak terkonjugasi melebihi 340 mol/l, ia melewati sawar darah otak dan menyebabkan kerusakan otak.
2. Ikterus hepatoseluler (hati)
Ikterus hepatoseluler (hati) disebabkan oleh kerusakan hepatosit dan kapiler empedu, misalnya pada infeksi virus akut, hepatitis kronis dan toksik.
Alasan peningkatan konsentrasi bilirubin dalam darah adalah kekalahan dan nekrosis sebagian sel hati. Ada keterlambatan dalam bilirubin di hati, yang difasilitasi oleh melemahnya tajam proses metabolisme pada hepatosit yang terkena, yang kehilangan kemampuan untuk melakukan berbagai fungsi biokimia dan fisiologis secara normal, khususnya, mentransfer bilirubin terkonjugasi (langsung) dari sel ke empedu melawan gradien konsentrasi. Untuk ikterus hepatoselular, merupakan karakteristik bahwa alih-alih diglucuronides bilirubin yang biasanya berlaku, terutama monoglucuronides terbentuk di sel hati yang terkena.
(Gbr. 13-17).
Akibat destruksi parenkim hepar, bilirubin direk yang dihasilkan sebagian masuk ke lingkaran besar sirkulasi, menyebabkan penyakit kuning. Ekskresi empedu juga terganggu. Bilirubin memasuki usus kurang dari biasanya.
Dengan ikterus hepatoseluler, konsentrasi dalam darah baik bilirubin total dan kedua fraksinya - tidak terkonjugasi (tidak langsung) dan terkonjugasi (langsung) meningkat.
Karena lebih sedikit bilirubinglucuronide yang masuk ke usus, jumlah urobilinogen yang terbentuk juga berkurang. Oleh karena itu, feses bersifat hipokolik, yaitu kurang berwarna. Sebaliknya, urin memiliki warna yang lebih intens karena adanya tidak hanya urobilin, tetapi juga bilirubin terkonjugasi, yang sangat larut dalam air dan diekskresikan dalam urin.
3. Ikterus mekanis atau obstruktif (sub-malam)
Mekanis, atau obstruktif (sub-hepatik-malam), penyakit kuning berkembang ketika ada pelanggaran sekresi empedu di usus duabelas jari. Hal ini dapat disebabkan oleh penyumbatan saluran empedu, seperti penyakit batu empedu, tumor pankreas, kantong empedu, hati, duodenum, peradangan kronis pankreas atau penyempitan saluran empedu umum pasca operasi (Gbr. 13-18).
Dengan penyumbatan total saluran empedu, bilirubin terkonjugasi dalam komposisi

Beras. 13-17. Pelanggaran siklus bilirubin-urobilinogen pada ikterus hepatoseluler. Di hati, laju reaksi glukuronidasi bilirubin berkurang (4), oleh karena itu, konsentrasi bilirubin tidak langsung dalam darah meningkat; karena pelanggaran parenkim hati, bagian dari bilirubinglucuronide yang terbentuk di hati memasuki darah (12) dan kemudian dikeluarkan dari tubuh dengan urin (10). Urobilins dan bilirubinglucuronides hadir dalam urin pasien. Angka-angka yang tersisa sesuai dengan tahapan metabolisme bilirubin pada Gambar. 13-16.
empedu tidak masuk ke usus, meskipun hepatosit terus memproduksinya. Karena bilirubin tidak masuk ke usus, tidak ada produk katabolisme urobilinogen dalam urin dan feses. Kotoran berubah warna. Karena jalur normal ekskresi bilirubin diblokir, ia bocor ke dalam darah, sehingga konsentrasi bilirubin terkonjugasi dalam darah pasien meningkat. Bilirubin terlarut diekskresikan dalam urin, memberikan warna oranye-coklat yang kaya.
B. DIAGNOSIS BANDING JAUNDICE
Saat mendiagnosis penyakit kuning, harus diingat bahwa dalam praktiknya, penyakit kuning jenis apa pun jarang dicatat dalam bentuk "murni". Kombinasi dari satu jenis atau lainnya lebih umum. Jadi, dengan penyakit kuning hemolitik yang parah, disertai dengan peningkatan konsentrasi bilirubin tidak langsung, berbagai organ pasti menderita, termasuk hati, yang dapat memasukkan unsur-unsur.

Beras. 13-18. Pelanggaran siklus bilirubin-urobilinogen pada ikterus obstruktif. Karena obstruksi kandung empedu, bilirubinglucuronide tidak disekresikan ke dalam empedu (5); kurangnya bilirubin di usus menyebabkan perubahan warna tinja (6); bilirubinglukuronida terlarut diekskresikan oleh ginjal dalam urin (10). Tidak ada urobilin dalam urin; bilirubin glukuronida yang terbentuk di hati memasuki aliran darah (12), akibatnya kandungan bilirubin langsung meningkat. Angka-angka yang tersisa sesuai dengan tahapan metabolisme bilirubin pada Gambar. 13-16.
ikterus parenkim, yaitu peningkatan bilirubin direk dalam darah dan urin. Pada gilirannya, ikterus parenkim, sebagai suatu peraturan, mencakup unsur-unsur mekanis. Dengan penyakit kuning subhepatik (mekanis), misalnya, dengan kanker kepala pankreas, peningkatan hemolisis tidak dapat dihindari sebagai akibat dari keracunan kanker dan, sebagai akibatnya, peningkatan bilirubin langsung dan tidak langsung dalam darah.
Jadi, hiperbilirubinemia dapat menjadi hasil dari kelebihan baik yang terikat maupun yang bebas
bilirubin. Pengukuran konsentrasi mereka secara terpisah diperlukan saat membuat diagnosis penyakit kuning. Jika konsentrasi bilirubin plasma<100 мкмоль/л и другие тесты функции печени дают нормальные результаты, возможно предположить, что повышение обусловлено за счёт непрямого билирубина. Чтобы подтвердить это, можно сделать анализ мочи, поскольку при повышении концентрации непрямого билирубина в плазме прямой билирубин в моче отсутствует.
Dalam diagnosis banding penyakit kuning, perlu memperhitungkan kandungan urobilinogen dalam urin. Biasanya, sekitar 4 mg urobilinogen diekskresikan dari tubuh dalam komposisi urin per hari. Jika peningkatan jumlah urobilinogen diekskresikan dalam urin, maka ini adalah bukti gagal hati, misalnya, dengan penyakit kuning hati atau hemolitik. Kehadiran dalam urin tidak hanya urobilinogen, tetapi juga bilirubin langsung menunjukkan kerusakan hati dan pelanggaran aliran empedu ke usus.
B. GANGGUAN METABOLISME BILIRUBIN
Beberapa penyakit diketahui di mana penyakit kuning disebabkan oleh gangguan metabolisme bilirubin yang diturunkan.
Sekitar 5% dari populasi didiagnosis dengan penyakit kuning herediter yang disebabkan oleh kelainan genetik pada struktur protein dan enzim yang bertanggung jawab untuk pengangkutan (penangkapan) bilirubin tidak langsung ke hati dan konjugasinya dengan asam glukuronat. Patologi ini diturunkan secara autosomal dominan. Dalam darah pasien, konsentrasi bilirubin tidak langsung meningkat.
Ada 2 jenis penyakit kuning herediter yang disebabkan oleh pelanggaran reaksi glukuronidasi di hati - pembentukan bilirubin langsung.
Tipe pertama ditandai dengan tidak adanya UDP-glucuronyltransferase. Sakit-
nie diwariskan secara resesif autosomal. Pengenalan fenobarbital, penginduksi UDP-glucuronyltransferase, tidak menyebabkan penurunan kadar bilirubin. Anak-anak meninggal pada usia dini karena perkembangan ensefalopati bilirubin.
Tipe kedua ditandai dengan penurunan aktivitas (defisiensi) UDP-glucuronyl transferase, hiperbilirubinemia terjadi karena bilirubin tidak langsung. Penyakit kuning merespon dengan baik terhadap pengobatan dengan fenobarbital.
Pelanggaran transpor aktif bilirubin glukuronida yang terbentuk di sel hati ke dalam empedu adalah karakteristik ikterus, yang diturunkan secara autosomal dominan. Ini dimanifestasikan oleh hiperbilirubinemia karena bilirubin langsung dan bilirubinuria (bilirubin langsung ditentukan dalam urin).
Hiperbilirubinemia familial pada bayi baru lahir dikaitkan dengan adanya inhibitor kompetitif konjugasi bilirubin (estrogen, asam lemak bebas) dalam ASI. Saat menyusui, inhibitor konjugasi bilirubin ditemukan dalam serum darah bayi. Hiperbilirubinemia semacam itu disebut sementara. Hiperbilirubinemia menghilang ketika anak dipindahkan ke makanan buatan. Hiperbilirubinemia yang tidak diobati menyebabkan perkembangan ensefalopati bilirubin dan kematian dini.