
Osteodistrofi herediter Albright. Pseudohipoparatiroidisme (osteodistrofi herediter Albright): kesulitan pencarian diagnostik diferensial. Pengamatan Klinis Gejala Pseudohipoparatiroidisme
Pseudohipoparatiroidisme
Apa itu Pseudohipoparatiroidisme -
Pseudohipoparatiroidisme(Nama samaran Yunani salah + hipoparatiroidisme; sinonim: Osteodistrofi herediter Albright, penyakit Albright) adalah penyakit keturunan yang langka Sistem Kerangka, mensimulasikan hipoparatiroidisme dan ditandai dengan gangguan metabolisme kalsium dan fosfor; sering disertai dengan keterlambatan perkembangan mental dan fisik.
Apa yang memprovokasi / Penyebab Pseudohipoparatiroidisme:
Penyebab pseudohipoparatiroidisme adalah kelainan bawaan - ketidakpekaan jaringan perifer terhadap aksi PTH.
Patogenesis (apa yang terjadi?) pada Pseudohipoparatiroidisme:
Dipercaya bahwa pseudohipoparatiroidisme didasarkan pada resistensi ginjal dan kerangka yang ditentukan secara genetik terhadap kerja hormon paratiroid sebagai akibat dari cacat pada sitoreseptor spesifik - hormon paratiroid - kompleks adenilat siklase, yang mengganggu pembentukan siklik 3 di ginjal. ", 5"-AMP, yang merupakan mediator intraseluler dari kerja hormon paratiroid pada proses metabolisme . Pseudohipoparatiroidisme adalah penyakit yang heterogen secara genetik. Pada beberapa pasien, sitoreseptor yang mengikat hormon paratiroid itu sendiri rusak (pseudohipoparatiroidisme tipe Ia); pada pasien lain, ada cacat pada protein pengikat nukleotida yang terlokalisasi di lapisan ganda lipid membran sel dan secara fungsional mengikat reseptor ke adenilat siklase (tipe Ib pseudohipoparatiroidisme). Beberapa pasien mengalami kekurangan enzim adenilat siklase itu sendiri (pseudohipoparatiroidisme tipe II). Defisiensi cAMP akibat cacat ini menyebabkan terganggunya sintesis protein spesifik yang menentukan efek biologis hormon paratiroid. Dengan demikian, sensitivitas organ target, khususnya ginjal, terhadap hormon paratiroid menjadi hilang. Akibatnya, ekskresi fosfor dalam urin menurun, terjadi hiperfosfatemia, dan hipokalsemia terjadi secara sekunder. Karena kelenjar paratiroid masih utuh pada pseudohipoparatiroidisme, kelenjar paratiroid sekunder dapat berkembang sebagai respons terhadap hipokalsemia, yang merangsang produksi hormon paratiroid. hiperparatiroidisme. Peningkatan pembentukan hormon paratiroid tidak menyebabkan peningkatan ekskresi fosfor dan cAMP melalui urin karena resistensi tubulus ginjal terhadap hormon paratiroid yang ditentukan secara genetik, tetapi disertai dengan perubahan jaringan tulang, karakteristik hiperparatiroidisme, yang menunjukkan terjaganya sensitivitas normal osteoklas terhadap hormon paratiroid. Dalam aktivitas pseudohipoparatiroidisme alkali fosfatase dalam serum darah meningkat atau dalam batas normal (0,5-1,3 μmol fosfor anorganik per 1 ml serum darah untuk 1 H inkubasi pada suhu 37°; definisi menurut Bodansky). Semua varian pseudohipoparatiroidisme merupakan penyakit keturunan, sifat pewarisannya adalah autosomal dominan. Rendahnya kesuburan pria yang menderita pseudohipoparatiroidisme menjelaskan jarangnya penularan penyakit ini dari ayah ke anak; wanita 2 kali lebih sering sakit dibandingkan pria.
Biasanya, dengan pseudohipoparatiroidisme, ditemukan hiperplasia kompensasi kelenjar paratiroid(keberadaan adenoma di dalamnya tidak khas). Perubahan khas hiperparatiroidisme dicatat pada jaringan tulang - osteoporosis difus, munculnya kista (disebut tumor coklat, tumor sel raksasa). Kalsium yang dilepaskan dari tulang disimpan dalam bentuk kalsifikasi di jaringan subkutan, serta di ginjal, otot, miokardium, dinding arteri besar, konjungtiva mata dan di sepanjang pinggiran kornea.
Gejala Pseudohipoparatiroidisme:
Tanda-tanda klinis pseudohipoparatiroidisme mirip dengan gejala idiopatik hipoparatiroidisme. Ada serangan kejang tonik yang terjadi secara spontan atau di bawah pengaruh bahan iritan. Kalsifikasi pada jaringan subkutan cenderung mengalami ulserasi. Osifikasi subkutan seringkali parah hingga menyerupai myositis ossificans. Ciri-cirinya antara lain keterbelakangan mental, pertumbuhan terhambat, wajah bulan, obesitas, dan brakidaktili, terutama pemendekan metakarpal dan metatarsal pertama, keempat, dan kelima. Eksostosis multipel, diskondroplasia, dan manifestasinya dapat diamati hiperparatiroidisme sekunder berupa resorpsi subperiosteal pada tulang jari; perubahan epifisis tulang sama seperti pada osteodisplasia fibrosa. Muntah sering dicatat, serta hematuria akibat pembentukan batu oksalat di saluran kemih, katarak lentikular, dan hipoplasia email gigi terdeteksi.
Pada pasien dengan pseudohipoparatiroidisme, seiring dengan penurunan sensitivitas organ target terhadap hormon paratiroid, resistensi terhadap hormon lain yang bergantung pada sistem adenilat siklase dapat diamati, misalnya gonad terhadap hormon gonadotropik, kelenjar tiroid terhadap hormon perangsang tiroid. , menargetkan organ untuk glukagon dan hormon antidiuretik. Ada peningkatan frekuensi penyakit autoimun Dan diabetes mellitus, hipotiroidisme dan hipertiroidisme diamati.
Ada juga pseudopseudohipoparatiroidisme, yang ditandai dengan tidak adanya hipokalsemia, hiperfosfatemia, kejang, dan osteomalasia.
Diagnosis Pseudohipoparatiroidisme:
Diagnosis pada kasus khas penyakit ini dibuat pada anak usia 5-10 tahun berdasarkan gambaran klinis yang khas, beberapa anomali dalam perkembangan kerangka tulang, adanya hipokalsemia, hiperfosfatemia, normal atau peningkatan aktivitas alkali fosfatase. dalam serum darah, penurunan ekskresi kalsium dan fosfor dalam urin, konten tinggi hormon paratiroid dalam darah. Adanya resistensi tubulus ginjal terhadap hormon paratiroid dikonfirmasi dengan tes berdasarkan penentuan jumlah fosfat dan cAMP yang diekskresikan dalam urin. Tidak adanya peningkatan signifikan kandungan fosfat dan cAMP dalam urin setelah pemberian hormon paratiroid kepada pasien menunjukkan resistensi ginjal terhadap kerja hormon paratiroid. Sebaliknya, pada pasien dengan hipoparatiroidisme idiopatik dan pasca operasi, setelah pemberian intravena 200 unit hormon paratiroid dalam urin, kandungan fosfat dan cAMP dalam 4 H meningkat 2-10 kali lipat dibandingkan garis dasar. Ekskresi hidroksiprolin melalui urin pada pasien pseudohipoparatiroidisme yang tidak diobati adalah normal atau sedikit meningkat, dan pada hipoparatiroidisme menurun. Diagnosis sinar-X pseudohipoparatiroidisme didasarkan pada identifikasi perubahan spesifik pada tulang dan jaringan lunak.
Pseudohipoparatiroidisme yang dikombinasikan dengan hipogonadisme pada wanita harus dibedakan Shereshevsky - Sindrom Turner, yang mana pseudopseudohipoparatiroidisme secara fenotip serupa. Pada sindrom Shereshevsky-Turner, kromatin seks tidak ada, di lokasi ovarium terdapat untaian jaringan ikat yang tidak terdeteksi selama pemeriksaan dubur dan USG.
Pengobatan Pseudohipoparatiroidisme:
Pengobatan hipokalsemia terdiri dari pemberian suplemen kalsium dalam dosis yang cukup untuk mempertahankan konsentrasi kalsium normal dalam darah. Sangat penting mendapat terapi vitamin D. Dosis awal dihitung mulai 2000 IU/ kg berat badan per hari, tetapi tidak lebih dari 100.000 IU per hari. Untuk menghindari overdosis sediaan vitamin D, perlu dilakukan pemantauan konsentrasi kalsium dalam darah setiap 3-7 hari selama dua minggu pertama pengobatan dan setiap bulan selama 2-3 bulan berikutnya. Setelah konsentrasi kalsium yang stabil dalam darah tercapai, cukup dilakukan pemeriksaan setiap 2-3 bulan sekali. Anda bisa menggunakan kalsitrin, dihydrotachysterol, oxydevit, serta obat lain bentuk aktif vitamin D. Diet terbatas fosfor membantu menormalkan konsentrasi kalsium dalam darah dan menghilangkan gejala hiperparatiroidisme sekunder. Jika kelenjar lain tidak mencukupi sekresi internal melakukan terapi penggantian dengan hormon yang sesuai. Pengobatan dengan hormon paratiroid tidak efektif. Untuk menghentikan serangan kejang, larutan 10% kalsium klorida atau kalsium glukonat diberikan secara intravena; secara oral - larutan kalsium klorida 5-10%, 1 sendok makan 3-4 kali sehari: kalsium glukonat, kalsium laktat - hingga 10 G dalam sehari.
Prognosis dengan terapi rasional baik. Mengingat sifat pseudohipoparatiroidisme yang bersifat herediter, maka diperlukan konseling genetik medis mengenai kemungkinan terjadinya pseudohipoparatiroidisme pada keturunannya.
Pencegahan Pseudohipoparatiroidisme:
Dokter mana yang harus Anda hubungi jika Anda menderita Pseudohipoparatiroidisme:
Apakah ada sesuatu yang mengganggumu? Ingin tahu informasi lebih lengkap tentang Pseudohipoparatiroidisme, Penyebabnya, Gejalanya, Cara Pengobatan dan Pencegahannya, Perjalanan Penyakit dan Pola Makan Setelahnya? Atau apakah Anda memerlukan pemeriksaan? Kamu bisa membuat janji dengan dokter– klinik Eurolaboratorium selalu siap melayani Anda! Dokter terbaik akan memeriksa dan mempelajari Anda tanda-tanda eksternal dan akan membantu Anda mengidentifikasi penyakit berdasarkan gejalanya, memberi saran dan memberikan bantuan yang diperlukan serta membuat diagnosis. kamu juga bisa panggil dokter di rumah. Klinik Eurolaboratorium terbuka untuk Anda sepanjang waktu.
Cara menghubungi klinik:
Nomor telepon klinik kami di Kyiv: (+38 044) 206-20-00 (multi-channel). Sekretaris klinik akan memilih hari dan waktu yang tepat bagi Anda untuk mengunjungi dokter. Koordinat dan arah kami ditunjukkan. Lihatlah lebih detail tentang semua layanan klinik di dalamnya.
(+38 044) 206-20-00
Jika Anda sebelumnya pernah melakukan penelitian apa pun, Pastikan untuk membawa hasilnya ke dokter untuk konsultasi. Jika penelitian belum dilakukan, kami akan melakukan segala sesuatu yang diperlukan di klinik kami atau dengan rekan kami di klinik lain.
Anda? Penting untuk mengambil pendekatan yang sangat hati-hati terhadap kesehatan Anda secara keseluruhan. Masyarakat kurang memberikan perhatian gejala penyakit dan tidak menyadari bahwa penyakit tersebut dapat mengancam nyawa. Banyak sekali penyakit yang awalnya tidak muncul di tubuh kita, namun pada akhirnya ternyata sudah terlambat untuk mengobatinya. Setiap penyakit memiliki gejala dan ciri khasnya masing-masing manifestasi eksternal- disebut demikian gejala penyakit. Mengidentifikasi gejala merupakan langkah awal dalam mendiagnosis penyakit secara umum. Untuk melakukan ini, Anda hanya perlu melakukannya beberapa kali dalam setahun. diperiksakan ke dokter untuk tidak hanya mencegah penyakit yang mengerikan, tetapi juga untuk menjaga kesehatan jiwa pada tubuh dan organisme secara keseluruhan.
Jika Anda ingin bertanya kepada dokter, gunakan bagian konsultasi online, mungkin Anda akan menemukan jawaban atas pertanyaan Anda di sana dan membacanya tips perawatan diri. Jika Anda tertarik dengan ulasan tentang klinik dan dokter, coba cari informasi yang Anda butuhkan di bagian tersebut. Daftar juga di portal medis Eurolaboratorium untuk tetap mendapatkan informasi terbaru berita terbaru dan update informasi pada website yang secara otomatis akan dikirimkan kepada Anda melalui email.
Penyakit lain dari golongan Penyakit sistem endokrin, gangguan gizi dan gangguan metabolisme :
| Krisis Addisonian (insufisiensi adrenal akut) |
| Adenoma payudara |
| Distrofi adiposogenital (penyakit Perchkranz-Babinski-Fröhlich) |
| Sindrom adrenogenital |
| Akromegali |
| Kegilaan nutrisi (distrofi nutrisi) |
| Alkalosis |
| Alkaptonuria |
| Amiloidosis (distrofi amiloid) |
| Amiloidosis lambung |
| Amiloidosis usus |
| Amiloidosis pulau pankreas |
| Amiloidosis hati |
| Amiloidosis esofagus |
| Asidosis |
| Malnutrisi energi protein |
| Penyakit sel I (mukolipidosis tipe II) |
| Penyakit Wilson-Konovalov (distrofi hepatoserebral) |
| Penyakit Gaucher (lipidosis glukoserebrosida, glukoserebrosidosis) |
| Penyakit Itsenko-Cushing |
| Penyakit Krabbe (leukodistrofi sel globoid) |
| Penyakit Niemann-Pick (sphingomyelinosis) |
| penyakit Fabry |
| Gangliosidosis GM1 tipe I |
| Gangliosidosis GM1 tipe II |
| Gangliosidosis GM1 tipe III |
| Gangliosidosis GM2 |
| Gangliosidosis GM2 tipe I (kebodohan luar biasa dari penyakit Tay-Sachs, Tay-Sachs) |
| Gangliosidosis GM2 tipe II (penyakit Sandhoff, kebodohan amaurotic Sandhoff) |
| Gangliosidosis GM2 remaja |
| Gigantisme |
| Hiperaldosteronisme |
| Hiperaldosteronisme sekunder |
| Hiperaldosteronisme primer (sindrom Conn) |
| Hipervitaminosis D |
| Hipervitaminosis A |
| Hipervitaminosis E |
| Hipervolemia |
| Koma hiperglikemik (diabetes). |
| Hiperkalemia |
| Hiperkalsemia |
| Hiperlipoproteinemia tipe I |
| Hiperlipoproteinemia tipe II |
| Hiperlipoproteinemia tipe III |
| Hiperlipoproteinemia tipe IV |
| Hiperlipoproteinemia tipe V |
| Koma hiperosmolar |
| Hiperparatiroidisme sekunder |
| Hiperparatiroidisme primer |
| Hiperplasia timus (kelenjar timus) |
| Hiperprolaktinemia |
| Hiperfungsi testis |
| Hiperkolesterolemia |
| Hipovolemia |
| Koma hipoglikemik |
| Hipogonadisme |
| Hipogonadisme hiperprolaktinemia |
| Hipogonadisme terisolasi (idiopatik) |
| Hipogonadisme kongenital primer (anorkisme) |
| Hipogonadisme didapat primer |
| Hipokalemia |
| Hipoparatiroidisme |
| Hipopituitarisme |
| Hipotiroidisme |
| Glikogenosis tipe 0 (aglikogenosis) |
| Glikogenosis tipe I (penyakit Gierke) |
| Glikogenosis tipe II (penyakit Pompe) |
| Glikogenosis tipe III (Penyakit campak, penyakit Forbes, batas dekstrinosis) |
| Glikogenosis tipe IV (penyakit Andersen, amilopektinosis, glikogenosis difus dengan sirosis hati) |
| Glikogenosis tipe IX (penyakit Haga) |
| Glikogenosis tipe V (penyakit McArdle, defisiensi miofosforilase) |
| Glikogenosis tipe VI (Penyakitnya, defisiensi hepatofosforilase) |
| Glikogenosis tipe VII (penyakit Tarui, defisiensi miofosfofruktokinase) |
| Glikogenosis tipe VIII (penyakit Thomson) |
| Glikogenosis tipe XI |
| Glikogenosis tipe X |
| Kekurangan (kekurangan) vanadium |
| Defisiensi magnesium (kekurangan) |
| Defisiensi mangan (kekurangan) |
| Defisiensi tembaga (kekurangan) |
| Kekurangan (kekurangan) molibdenum |
| Kekurangan (kekurangan) kromium |
| Kekurangan zat besi |
| Defisiensi kalsium (kekurangan kalsium gizi) |
| Defisiensi zinc (kekurangan zinc dalam makanan) |
| Koma ketoasidosis diabetik |
| Disfungsi ovarium |
| Gondok difus (endemik). |
| Pubertas tertunda |
| Estrogen berlebih |
| Involusi kelenjar susu |
| Dwarfisme (perawakan pendek) |
| Kwashiorkor |
| Mastopati kistik |
| Xantinuria |
| Koma asam laktat |
| Leucinosis (penyakit sirup maple) |
| Lipidosis |
| Lipogranulomatosis jauh |
| Lipodistrofi (degenerasi lemak) |
| Lipodistrofi umum bawaan (sindrom Seyp-Lawrence) |
| Lipodistrofi hipermuskular |
| Lipodistrofi pasca injeksi |
| Lipodistrofi segmental progresif |
| lipomatosis |
| Lipomatosis itu menyakitkan |
| Leukodistrofi metakromatik |
| Koma miksedema |
Pseudohipoparatiroidisme - cukup penyakit langka sistem kerangka, yang intinya adalah pelanggaran metabolisme kalsium dan fosfor. Penderita biasanya mengalami terhambatnya perkembangan mental dan fisik. Penyakit ini bersifat keturunan.
Dari fakta bahwa nama penyakit ini memiliki awalan “semu”, dapat dengan mudah dipahami bahwa penyakit ini meniru hipoparatiroidisme. Nama kedua penyakit ini adalah osteodistrofi herediter Albright, dinamai menurut nama dokter yang mempelajari dan mendeskripsikan patologi ini pada pertengahan abad terakhir.
Jenis pseudohipoparatiroidisme
Ada dua jenis pseudohipoparatiroidisme - tergantung pada apakah kadar kalsium dalam darah berubah atau tidak. Jenis penyakit Albright yang pertama juga sama Tanda-tanda klinis, seperti pada hipoparatiroidisme idiopatik, tetapi ditandai dengan penurunan kadar kalsium dalam darah. Tidak ada sensitivitas jaringan terhadap hormon paratiroid. Sebaliknya, jenis pseudohipoparatiroidisme yang kedua berbeda karena kadar kalsiumnya normal. Oleh karena itu, jenis ini disebut pseudopseudohipoparatiroidisme. Ngomong-ngomong, menurut ahli endokrinologi, satu bentuk dapat dengan mudah berubah menjadi bentuk lain, dan anggota keluarga yang sama mungkin mengalaminya jenis yang berbeda penyakit.
Penyebab pseudohipoparatiroidisme
Gangguan metabolisme fosfor-kalsium berkembang karena resistensi jaringan terhadap hormon paratiroid yang diproduksi oleh kelenjar paratiroid. Berdasarkan pseudohipoparatiroidisme, fenomena gangguan sensitivitas jaringan terhadap hormon yang diproduksi oleh kelenjar endokrin atau diberikan secara eksternal telah dikonfirmasi untuk pertama kalinya.
Pseudohipoparatiroidisme adalah kelainan genetik. Hal ini disebabkan oleh sindrom bawaan - reseptor seluler spesifik - hormon paratiroid-adenilat siklase, yang menyebabkan jaringan perifer kehilangan sensitivitas terhadap hormon paratiroid.
Siapa yang paling rentan terkena pseudohipoparatiroidisme? Pertama-tama, anak-anak pasien dan kerabat lainnya, karena penyakit ini bersifat herediter, autosomal dominan. Wanita lebih sering menderita penyakit Albright. Para ahli menjelaskan hal ini dengan fakta bahwa pria dengan pseudohipoparatiroidisme memiliki kesuburan yang rendah, dan faktor keturunan itu sendiri diturunkan dari ayah ke anak hanya dalam kasus-kasus tertentu.
Tidak ada statistik mengenai seberapa umum penyakit ini, sekitar tiga ratus kasus dijelaskan dalam karya medis.
Gejala pseudohipoparatiroidisme
- Masalah organ sistem endokrin dengan pseudohipoparatiroidisme, gejala utama juga ditentukan: pasien biasanya kekar, lebih pendek dari yang lain, karena memendek anggota tubuh bagian bawah, mengalami obesitas, kadar glukosa darah meningkat, wajah berbentuk bulan;
- Perwakilan dari jenis kelamin yang lebih adil terpengaruh siklus menstruasi. Seringkali tanda-tanda ini disertai dengan akromegali, ginekomastia, berbagai proses autoimun, sindrom Itsenko-Cushing dan penyakit lainnya. Pada pasien tersebut, risiko diabetes melitus dan diabetes insipidus juga lebih tinggi;
- Mereka yang menderita penyakit Albright sering mengeluhkan masalah pada giginya (enamelnya rusak); pasien juga terganggu oleh muntah-muntah, kejang-kejang tonik - baik yang spontan maupun yang timbul karena pengaruh iritan; urin dapat diamati dalam darah. Ada risiko katarak. Ditandai dengan keterbelakangan mental. Anak-anak dengan penyakit ini tidak dapat mengikuti kurikulum sekolah, daya ingatnya menurun;
- DI DALAM sistem osteoartikular perubahan signifikan juga terjadi - osteoporosis difus muncul, kista (tumor coklat) berkembang. Pasien dengan pseudohipoparatiroidisme lebih mungkin menderita patah tulang atau kelainan bentuk tulang. Tulang metakarpal dan metatarsal pertama, keempat dan kelima memendek, dan tanda-tanda resorpsi tulang terlihat pada tangan dan kaki. Selain itu, kalsium yang dikeluarkan dari tulang mulai menumpuk di jaringan subkutan, membentuk kalsifikasi (ini juga terjadi pada myositis ossificans), di area persendian, dan rongga tengkorak. Kalsifikasi juga terdapat pada otot, ginjal, miokardium dan pada dinding arteri besar;
- Lain tanda Penyakit Albright - pigmentasi kulit. Bintik-bintik coklat menyebar ke seluruh tubuh (baik di satu sisi maupun di sisi lainnya).
Mengenai kemampuan bekerja, tergantung pada beratnya proses dan efisiensi perawatan obat. Jika bentuk penyakitnya laten dan tidak ada serangan yang jelas, maka kemampuan bekerja sebagian tetap terjaga. Namun ada sejumlah batasan - khususnya, Anda tidak dapat bekerja dengan mekanisme bergerak atau dalam transportasi; ketegangan neuropsikik yang berlebihan dan kerja fisik yang berlebihan juga dilarang. Jika sering terjadi serangan kejang, keterbelakangan mental yang jelas terlihat, atau gangguan penglihatan yang signifikan akibat katarak, maka pasien tersebut sudah dianggap cacat.
Diagnosis pseudohipoparatiroidisme
Gejalanya cukup spesifik dan jika mulai muncul, sebaiknya segera hubungi dokter yang berkualifikasi. Ini adalah awal dan diagnosa yang benar menjamin keberhasilan pengobatan.
Karena penyakit Albright bersifat bawaan, biasanya pasien didiagnosis pada masa kanak-kanak (prasekolah, junior usia sekolah). Dokter genetika membuat kesimpulan berdasarkan gejala klinis, dan metode diagnostik laboratorium dan instrumental juga digunakan.
Secara khusus, untuk memperjelas diagnosis, pasien mendonorkan darah (pada pasien dengan pseudohipoparatiroidisme, kadar kalsium rendah, fosfor dan hormon paratiroid tinggi, aktivitas alkaline fosfatase dalam darah tinggi), urin (pada pasien, ini analisis akan menunjukkan penurunan fosfor dan kalsium yang diekskresikan). Tes khusus juga dilakukan untuk menunjukkan seberapa sensitif jaringan tubulus ginjal pasien terhadap hormon paratiroid. Terkadang perlu untuk menilai tingkat hidroksiprolin.
Metode instrumental melibatkan diagnostik sinar-X (paling informatif ketika mengidentifikasi perubahan spesifik pada tulang dan jaringan otot).
Pengobatan pseudohipoparatiroidisme
Patologi keturunan yang langka ini diobati dengan suplemen kalsium. Dokter memilih dosis berdasarkan kandungan kalsium yang dibutuhkan untuk menjaga homeostasis. Dan agar kalsium dapat diserap tanpa masalah, sediaan vitamin D juga termasuk dalam rejimen terapi.Selain itu, dokter untuk pasien pseudohipoparatiroidisme akan mengembangkan pola makan yang membatasi jumlah makanan yang mengandung fluoride. Diet ini akan membantu menormalkan konsentrasi kalsium dalam darah dan menghilangkan tanda-tanda hiperparatiroidisme sekunder.
Jika penyakit Albright disertai penyakit lain gangguan endokrin, maka mungkin diperlukan terapi penggantian hormon. Kejang-kejang diredakan dengan larutan kalsium klorida atau kalsium glukonat, yang diberikan secara intravena. Jika katarak muncul, pengobatannya dilakukan oleh dokter mata, dan psikolog membantu meringankan gangguan kognitif.
Dengan rejimen pengobatan yang dipikirkan dengan matang, prognosisnya sangat baik. Namun semua penderita pseudohipoparatiroidisme sebaiknya rutin berkonsultasi dengan ahli genetika guna mencegah penyakit tersebut pada keturunannya.
Pencegahan pseudohipoparatiroidisme
Mengingat penyakit ini bersifat keturunan satu-satunya jalan Pencegahan bagi mereka yang memiliki riwayat keluarga pseudohipoparatiroidisme adalah dengan mengunjungi dokter ahli genetika medis secara rutin untuk berkonsultasi.
(Penyakit Albright) adalah osteodistrofi herediter yang disebabkan oleh resistensi jaringan perifer terhadap hormon paratiroid, yang disertai dengan gangguan metabolisme kalsium-fosfor, keterlambatan perkembangan fisik dan mental. Pseudohipoparatiroidisme terjadi dengan gejala osteoporosis difus, kejang tonik, patah tulang dan deformasi tulang, pengendapan kalsium pada otot dan pembuluh darah, pembentukan batu pada saluran kemih, keterbelakangan pertumbuhan dan keterbelakangan mental. Untuk mendiagnosis pseudohipoparatiroidisme, kadar kalsium, alkali fosfatase, dan hormon paratiroid dalam darah ditentukan; ekskresi fosfor dan kalsium melalui urin; tes fungsional dengan diperkenalkannya hormon paratiroid; Diagnostik sinar-X. Pengobatan pseudohipoparatiroidisme melibatkan koreksi obat hipokalsemia.
ICD-10
E20.1

Informasi Umum
Pseudohipoparatiroidisme adalah kelainan herediter yang langka; Secara endokrinologi, hanya sekitar 300 kasus penyakit ini yang diketahui. Pseudohipoparatiroidisme dalam manifestasinya menyerupai hipoparatiroidisme. Namun, jika hipoparatiroidisme didasarkan pada defisiensi primer hormon paratiroid yang disebabkan oleh penurunan aktivitas fungsional kelenjar paratiroid, maka pada penyakit Albright, sindrom pseudohipoparatiroid disebabkan oleh pelanggaran sensitivitas jaringan target terhadap kerja paratiroid. hormon dengan tingkat sekresi yang cukup.
Pseudohipoparatiroidisme pertama kali dijelaskan pada tahun 1942. F. Albright dan menurut penulis disebut “penyakit Albright” atau “osteodistrofi herediter Albright”. Varian pertama penyakit ini ditandai dengan kelainan perkembangan tulang, resistensi jaringan perifer terhadap hormon paratiroid, hipokalsemia dan Gambaran klinis, mirip dengan hipoparatiroidisme idiopatik. Varian kedua penyakit Albright terjadi menurut tipe normokalsemik dan dalam literatur disebut pseudopseudohipoparatiroidisme. Kasus perkembangan varian penyakit normo dan hipokalsemik dalam satu keluarga, serta transisi dari satu bentuk ke bentuk lainnya, telah dijelaskan.
Penyebab pseudohipoparatiroidisme
Dalam varian pseudohipoparatiroidisme apa pun, penyakit ini bersifat herediter dengan jenis pewarisan autosomal dominan yang terkait dengan kromosom X. Sebuah studi tentang silsilah menunjukkan bahwa jumlah wanita dengan pseudohipoparatiroidisme 2 kali lebih tinggi dibandingkan jumlah pria yang sakit; Selain itu, penyakit Albright tidak menular dari ayah ke anak laki-lakinya.
Pseudohipoparatiroidisme disebabkan oleh resistensi genetik kerangka dan ginjal terhadap kerja hormon paratiroid. Hal ini disebabkan oleh kerusakan pada reseptor spesifik membran plasma sel target dan defisiensi enzim adenilat siklase, protein kinase, siklik 3, 5-adenosin monofosfat (AMP) di tubulus distal nefron, yang disertai dengan reabsorpsi fosfor. Penurunan ekskresi fosfor urin menyebabkan hiperfosfatemia dan hipokalsemia sekunder.
Karena pada pseudohipoparatiroidisme kelenjar paratiroid tetap utuh, hipokalsemia dapat menyebabkan stimulasi sekresi hormon paratiroid dan perkembangan hiperparatiroidisme sekunder. Dengan pseudohipoparatiroidisme, hiperplasia kompensasi kelenjar paratiroid biasanya terjadi (perkembangan adenoma tidak khas), perubahan jaringan tulang (osteoporosis, kista), pengendapan kalsifikasi di otot rangka, jaringan subkutan, serta di ginjal, dinding arteri, miokardium, konjungtiva dan kornea. Pseudohipoparatiroidisme sering dikombinasikan dengan hipertensi arteri, diabetes mellitus, arteritis, dan poliartritis.
Gejala pseudohipoparatiroidisme
Manifestasi klinis pseudohipoparatiroidisme mirip dengan hipoparatiroidisme idiopatik. Gejala endokrin pseudohipoparatiroidisme termasuk perawakan pendek, obesitas dan hiperglikemia, wajah bulan, dan oligomenore. Penyakit Albright sering dikombinasikan dengan kelainan endokrin lainnya - akromegali, sindrom Cushing, ginekomastia, hipotiroidisme, atau hipertiroidisme.
Tanda-tanda osteoartikular dari pseudohipoparatiroidisme termasuk brachydactyly dengan pemendekan metakarpal dan metatarsal ke-1, ke-4, dan ke-5; eksostosis, diskondroplasia, perubahan ujung epifisis tulang, resorpsi tulang jari. Ditandai dengan keterlambatan tumbuh gigi, hipoplasia email gigi, osifikasi subkutan.
Gejala neurologis pseudohipoparatiroidisme diwakili oleh serangan kejang tonik, yang dapat terjadi secara spontan atau di bawah pengaruh rangsangan apa pun. Untuk pasien dengan pseudohipoparatiroidisme, keterbelakangan mental merupakan ciri khasnya. Gejala lain dari pseudohipoparatiroidisme termasuk urolitiasis, hematuria, muntah, dan perkembangan katarak lentikular. Dengan pseudopseudohipoparatiroidisme, tidak ada hipokalsemia, hiperfosfatemia, osteomalacia, dan sindrom kejang.
Diagnosis pseudohipoparatiroidisme
Dalam kasus yang khas, pseudohipoparatiroidisme didiagnosis pada anak usia 5-10 tahun, dengan mempertimbangkan gejala khas, data laboratorium dan radiologi. Pada pasien dengan pseudohipoparatiroidisme, hipokalsemia, hiperfosfatemia, normal atau peningkatan aktivitas alkali fosfatase dalam darah, dan peningkatan kadar hormon paratiroid dalam serum darah ditentukan; penurunan ekskresi kalsium dan fosfor dalam urin.
Ketidakpekaan tubulus ginjal terhadap hormon paratiroid dikonfirmasi dengan tes berdasarkan penentuan jumlah fosfat dan siklik adenosin monofosfat yang diekskresikan dalam urin. Pada pseudohipoparatiroidisme sebagai respons terhadap pemberian intravena hormon paratiroid, tidak terjadi peningkatan signifikan kandungan fosfat dan cAMP dalam urin.
Dalam kasus pseudohipoparatiroidisme, koreksi hipokalsemia diperlukan, dan oleh karena itu terapi dengan sediaan kalsium dilakukan dalam dosis yang memungkinkan mempertahankan konsentrasi kalsium normal dalam darah. Asupan vitamin D dan bentuk aktifnya diindikasikan di bawah kendali konsentrasi kalsium dalam serum darah. Pengobatan dengan hormon paratiroid tidak efektif. Untuk menormalkan konsentrasi kalsium dan menghilangkan gejala hiperparatiroidisme sekunder, perlu dilakukan diet dengan asupan fosfor terbatas.
Jika terjadi insufisiensi fungsional kelenjar lain, terapi penggantian dengan hormon yang sesuai dilakukan. Jika kejang terjadi, pemberian larutan kalsium intravena diindikasikan.
Diagnosis tepat waktu dan terapi rasional pseudohipoparatiroidisme memungkinkan kita berbicara tentang prognosis positif seumur hidup dan kemungkinan mengendalikan perjalanan penyakit. Mengingat sifat genetik dari pseudohipoparatiroidisme, disarankan untuk melakukan konseling genetik medis untuk menilai risiko penyakit Albright pada keturunannya.
![]()
Keterangan:
Pseudohipoparatiroidisme (Yunani pseudēs false +; sinonim: osteodistrofi herediter Albright, penyakit Albright) adalah penyakit keturunan langka pada sistem kerangka, yang menyerupai hipoparatiroidisme dan ditandai dengan gangguan metabolisme kalsium dan fosfor; sering disertai dengan keterlambatan perkembangan mental dan fisik.
Gejala:
Tanda-tanda klinis pseudohipoparatiroidisme mirip dengan hipoparatiroidisme idiopatik. Ada serangan tonik yang terjadi secara spontan atau di bawah pengaruh rangsangan apa pun. Kalsifikasi pada jaringan subkutan cenderung mengalami ulserasi. Osifikasi subkutan seringkali parah hingga menyerupai ossificans. Ditandai dengan keterbelakangan mental, keterbelakangan pertumbuhan, wajah berbentuk bulan, dan terutama pemendekan tulang metakarpal dan metatarsal pertama, keempat dan kelima. Eksostosis multipel, diskondroplasia, dan manifestasi sekunder berupa resorpsi subperiosteal pada tulang jari dapat diamati; perubahan epifisis tulang sama seperti pada osteodisplasia fibrosa. Muntah sering dicatat, serta hematuria akibat pembentukan batu oksalat di saluran kemih, katarak lentikular, dan hipoplasia email gigi terdeteksi.
Pada pasien dengan pseudohipoparatiroidisme, seiring dengan penurunan sensitivitas organ target terhadap hormon paratiroid, resistensi terhadap hormon lain yang bergantung pada sistem adenilat siklase dapat diamati, misalnya gonad terhadap hormon gonadotropik, kelenjar tiroid terhadap hormon perangsang tiroid. , menargetkan organ untuk glukagon dan hormon antidiuretik. Ada peningkatan kejadian penyakit autoimun dan diabetes mellitus, dan diamati.
Ada juga pseudopseudohipoparatiroidisme, yang ditandai dengan tidak adanya kejang dan osteomalacia.
Penyebab:
Penyebab pseudohipoparatiroidisme adalah kelainan bawaan - ketidakpekaan jaringan perifer terhadap aksi PTH.
Perlakuan:
Untuk pengobatan, berikut ini ditentukan:
Pengobatan hipokalsemia terdiri dari pemberian suplemen kalsium dalam dosis yang cukup untuk mempertahankan konsentrasi kalsium normal dalam darah. Terapi vitamin D sangat penting.Dosis awal dihitung dari 2000 IU/kg berat badan per hari, tetapi tidak lebih dari 100.000 IU per hari. Untuk menghindari overdosis sediaan vitamin D, perlu dilakukan pemantauan konsentrasi kalsium dalam darah setiap 3-7 hari selama dua minggu pertama pengobatan dan setiap bulan selama 2-3 bulan berikutnya. Setelah konsentrasi kalsium yang stabil dalam darah tercapai, cukup dilakukan pemeriksaan setiap 2-3 bulan sekali. Anda dapat menggunakan kalsitrin, dihidrotakisterol, oksidavit, serta sediaan lain dari bentuk aktif vitamin D. Diet dengan pembatasan fosfor membantu menormalkan konsentrasi kalsium dalam darah dan menghilangkan gejala hiperparatiroidisme sekunder. Jika kelenjar endokrin lain tidak mencukupi, terapi penggantian dengan hormon yang sesuai dilakukan. Pengobatan dengan hormon paratiroid tidak efektif. Untuk menghentikan serangan kejang, larutan 10% kalsium klorida atau kalsium glukonat diberikan secara intravena; secara oral - larutan kalsium klorida 5-10%, 1 sendok makan 3-4 kali sehari: kalsium glukonat, kalsium laktat - hingga 10 g per hari.
Prognosis dengan terapi rasional baik. Mengingat sifat pseudohipoparatiroidisme yang bersifat herediter, maka diperlukan konseling genetik medis mengenai kemungkinan terjadinya pseudohipoparatiroidisme pada keturunannya.
EV. Tozliyan, ahli endokrinologi anak, ahli genetika, Ph.D., I.V. Shulyakova, ahli saraf, Ph.D.,
terpencil subdivisi struktural"Institut Penelitian Klinis Pediatri" Lembaga Pendidikan Anggaran Negara Pendidikan Profesi Tinggi "Universitas Kedokteran Riset Nasional Rusia dinamai N.I. Pirogov" dari Kementerian Kesehatan Federasi Rusia, Moskow
Kata kunci:
anak-anak, pseudohipoparatiroidisme, osteodistrofi herediter Albright, obesitas, hipokalsemia, diagnosis, resistensi terhadap hormon paratiroid.
Kata kunci: anak-anak, pseudohipoparatiroidisme, osteodistrofi herediter Albright, obesitas, hipokalsemia, diagnostik, resistensi hormon paratiroid.
Pseudohipoparatiroidisme (Yunani pseudes - palsu + hipoparatiroidisme; sinonim: osteodistrofi herediter Albright, sindrom ayam Jawa) adalah penyakit keturunan langka pada sistem kerangka, yang menyerupai hipoparatiroidisme dan ditandai dengan gangguan metabolisme kalsium dan fosfor; sering disertai dengan keterlambatan perkembangan mental dan fisik. Penyakit ini pertama kali dijelaskan oleh ahli endokrinologi Amerika Albright F. pada tahun 1942. Prevalensi penyakit ini adalah 7,9 per 1 juta orang.
DATA GENETIK
Pseudohipoparatiroidisme (PHP) adalah penyakit yang heterogen secara genetik. Data tentang jenis penularan herediter bertentangan: baik tipe X-linked dominan maupun autosomal dominan, resesif autosomal. Dalam kebanyakan kasus, perkembangan osteodistrofi herediter Albright dikaitkan dengan mutasi pada lokus 20q13 gen GNAS1 yang terletak pada kromosom 20 (Patten et al., 1990), yang mengkode protein Gs-alpha yang terkait dengan reseptor hormon paratiroid (PTH). . Fenotip serupa juga terdeteksi pada pasien dengan penghapusan interstisial lengan panjang kromosom 2 di lokus 2q37.
PATOGENESIS
Patogenesis pseudohipoparatiroidisme didasarkan pada resistensi ginjal dan kerangka yang ditentukan secara genetik terhadap kerja hormon paratiroid sebagai akibat dari kerusakan pada kompleks “sitoreseptor spesifik – hormon paratiroid – adenilat siklase”, yang mengganggu pembentukan siklik 3"- , 5"-adenosin monofosfat (cAMP) di ginjal, yang merupakan mediator intraseluler dari kerja hormon paratiroid pada proses metabolisme. Pseudohipoparatiroidisme bersifat genetik penyakit yang heterogen. Pada beberapa pasien, sitoreseptor itu sendiri yang mengikat hormon paratiroid rusak (pseudohipoparatiroidisme tipe 1A); yang lain memiliki cacat pada protein pengikat nukleotida, terlokalisasi di lapisan ganda lipid membran sel dan secara fungsional mengikat reseptor ke adenilat siklase (tipe 1B pseudohipoparatiroidisme). Beberapa pasien mengalami defisiensi enzim adenilat siklase itu sendiri (pseudohipoparatiroidisme tipe 2). Defisiensi cAMP akibat cacat ini menyebabkan terganggunya sintesis protein spesifik yang menentukan efek biologis hormon paratiroid. Dengan demikian, sensitivitas organ target terhadap hormon paratiroid hilang.
KARAKTERISTIK KLINIS
Saat ini ada 4 bentuk klinis patologi: tipe 1A, 1B, 1C dan 2. Pengetahuan tentang gambaran dan data klinis dan biokimianya penelitian genetik memungkinkan untuk diagnosis banding dalam bentuk nosologis itu sendiri.
Tanda-tanda umum yang memungkinkan seseorang untuk mencurigai penyakit ini adalah perkembangan fisik yang tidak proporsional, perawakan pendek (sampai dwarfisme) karena pemendekan anggota tubuh bagian bawah (foto 1), brachydactyly (foto 2), dan bentuk bulat “berbentuk bulan”. wajah (foto 3). Eksostosis dan aplasia gigi terkadang diamati.
Foto 1.
Penampilan anak dengan osteodistrofi Albright
(ciri-ciri fenotipe, perawakan pendek karena pemendekan anggota tubuh bagian bawah)
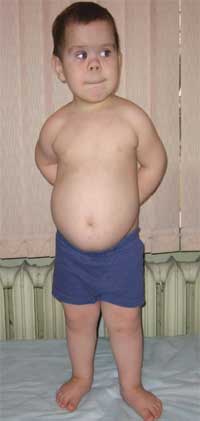
Foto 2.
Fitur sistem kerangka pasien
dengan osteodistrofi Albright
(brachydactyly - pemendekan jari)
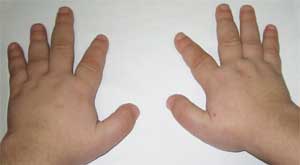
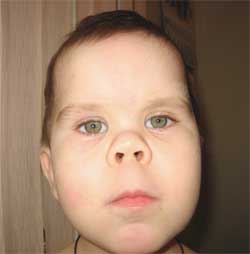
Foto 3.
Ciri-ciri fenotip anak
dengan osteodistrofi Albright
(wajah bulat berbentuk bulan)
Tanda patognomoniknya adalah pemendekan tajam tulang metakarpal dan metatarsal I, III dan V (terutama III dan IV), akibatnya jari-jari II pada tangan dan kaki lebih panjang dari yang lain, dan ketika tangan dikepalkan. mengepal, tidak ada tonjolan di area sendi metacarpophalangeal IV dan V - yang disebut brachymetaphalangism. Falang pendek dan lebar, penebalan kubah tengkorak dan demineralisasi tulang (osteoporosis), dan obesitas juga terdeteksi.
Keterbelakangan mental(biasanya tingkat keparahan sedang) ditemukan pada sekitar 20% pasien. Menurut beberapa penulis, keterbelakangan mental terjadi pada 70% kasus dengan hipokalsemia dan pada 30% kasus dengan normokalsemia. Proses mental pada pasien melambat. Dalam status neurologis, kecanggungan motorik dan reaksi neurotik sering dicatat: ketakutan, kecemasan, kegelisahan, mimpi buruk, peningkatan refleks, kejang yang bersifat tetanik dan disebabkan oleh hipokalsemia, terkadang kejang paroksismal. Gejala miopati juga telah dijelaskan: kelelahan otot, kelemahan otot. Gangguan ekstrapiramidal yang sering diamati: hiperkinesis koreiform, athetosis, hemispasme wajah, parkinsonisme, dalam beberapa kasus ada paroxysms epilepsi, gejala otak kecil: ataksia, kehilangan koordinasi.
Kalsifikasi jaringan lunak, kalsifikasi subkutan (dada, perut, tendon tumit) sering terdeteksi, dengan pemeriksaan histologis yang - osteoma kutis(Izraeli et al., 1992), otak (basal ganglia). Penting untuk diperhatikan bahwa kalsifikasi mungkin sudah ada sejak lahir. Akibat hipokalsemia, katarak biasanya berkembang dan terjadi kerusakan email gigi.
PSEUDOHIPOPARATROOSIS TIPE 1A
memiliki pola pewarisan autosomal dominan. Gen pseudohipoparatiroidisme tipe 1A - GNAS1 - terlokalisasi di lengan panjang kromosom 20, di lokus 20q13.2. Perkembangan penyakit ini dikaitkan dengan kekurangan protein pengikat nukleotida guanin (protein Gs). Pada saat yang sama, PTH, yang berikatan dengan reseptor jaringan target, tidak mampu mengaktifkan siklik adenosin monofosfat (cAMP) dan menyebabkan respons jaringan. Mungkin, mekanisme serupa mendasari perkembangan ketidakpekaan jaringan organ lain dan kelenjar endokrin (hipofungsi kelenjar tiroid, gonad, kelenjar pituitari, diabetes, serta berkurangnya respons hati terhadap pemberian glukagon), diamati pada pseudohipoparatiroidisme tipe 1A. Pada jenis patologi ini, peningkatan ekskresi cAMP yang normal dalam urin sebagai respons terhadap pemberian PTH eksogen tidak diamati. Penyakit ini lebih sering didiagnosis pada usia 5-10 tahun. Pasien memiliki perawakan pendek, leher pendek, wajah bulat, pemendekan tulang metakarpal dan metatarsal (biasanya pemendekan jari keempat dan lebih jarang jari kedua) - yang disebut brachy-metaphalangism. Kalsifikasi jaringan lunak dan kalsifikasi subkutan dicatat, yang dapat dideteksi saat lahir; keterlibatan simultan kelenjar endokrin lainnya sering diamati: kelenjar tiroid (hipofungsi), gonad, pankreas (diabetes mellitus). Akibat hipokalsemia, sering terjadi katarak dan kerusakan email gigi. Sebagai uji diagnostik diferensial untuk membedakan PHP tipe 1A dengan hipoparatiroidisme: tidak adanya efek klinis dari pemberian PTH parenteral berupa peningkatan kadar kalsium dalam darah dan peningkatan ekskresi fosfor ginjal melalui urin. (efek fosfat).
Pemeriksaan biokimia menunjukkan hipokalsemia, hiperfosfatemia, peningkatan kadar hormon paratiroid dalam darah, dan hipofosfaturia. Kadar protein Gs dalam darah berkurang. Pada pemeriksaan rontgen Sistem kerangka menunjukkan pemendekan tulang metakarpal dan metatarsal, demineralisasi umum, dan penebalan tulang kubah tengkorak.
PSEUDOHIPOPARATROOSIS TIPE 1B
memiliki tipe pewarisan autosomal dominan, namun tipe pewarisan dominan terkait X tidak dikecualikan. Penting untuk diingat bahwa kadang-kadang ada penetrasi gen penyakit yang tidak lengkap dan kemungkinan pembawa patologi yang tersembunyi. Oleh karena itu, pemeriksaan klinis (deteksi perjalanan penyakit subklinis) dan pemeriksaan biokimia (penentuan kadar kalsium, fosfor, PTH dalam darah) terhadap dugaan pembawa penyakit dianjurkan. PHP tipe 1B disebabkan oleh kekurangan reseptor jaringan terhadap hormon paratiroid pada organ target dan terbatasnya resistensi terhadap hormon paratiroid. Gambaran klinisnya mirip dengan tipe 1A, tetapi tidak ada kerusakan pada kelenjar endokrin lainnya, dan osteodistrofi lebih jarang terjadi.
Pasien tidak memiliki respon ginjal terhadap pemberian hormon paratiroid eksogen dalam bentuk peningkatan ekskresi siklik adenosin monofosfat dalam urin, namun, tidak seperti tipe 1A, kadar protein Gs dalam darah normal. Wanita lebih sering terkena dibandingkan pria, namun tingkat keparahan penyakit ini bisa sama baik pada pria maupun wanita.
PSEUDOHIPOPARATROOSIS TIPE 1C
beberapa penulis mengidentifikasinya dengan pseudo-pseudohypoparathyroidism (PPHP), dijelaskan oleh Albright F. pada tahun 1952. Hal ini ditandai dengan gambaran klinis khas PHP, namun kadar kalsium dan fosfor dalam darah dan urin tetap dalam batas normal. Kadar protein PTH dan Gs dalam darah juga tetap tidak berubah tingkat normal. Beberapa pasien dengan PHP tipe 1C mengalami penghapusan de novo pada kromosom 2. Kemungkinan varian penyakit tersebut merupakan subtipe dari PGP tipe 1A.
PSEUDOHIPOPARATROOSIS TIPE 2
secara klinis mirip dengan jenis penyakit lain, tetapi memiliki pola pewarisan autosomal resesif. Keberadaan bentuk patologi autosomal dominan tidak dapat dikesampingkan. Patogenesis perkembangan dikaitkan dengan resistensi intraseluler terhadap cAMP. PTH kemudian berikatan dengan reseptor dan menyebabkan respon seluler normal terhadap PTH berupa peningkatan ekskresi cAMP. Ketidakpekaan intraseluler terhadap cAMP, bagaimanapun, tidak memungkinkan efek PTH sepenuhnya terwujud. Pada saat yang sama, itu tetap ada reaksi biasa ginjal terhadap pemberian hormon paratiroid eksogen dalam bentuk peningkatan ekskresi siklik adenosin monofosfat dalam urin. Ada dugaan bahwa PHP tipe 2 mungkin berhubungan dengan kekurangan vitamin D.
Dengan demikian, jenis PGP yang teridentifikasi secara klinis ditandai dengan berkurangnya sensitivitas organ target terhadap PTH, namun berbeda dalam mekanisme patogenetik pembentukan ketidakpekaan jaringan.
DIAGNOSA
Tes diagnostik diferensial laboratorium dapat menjadi pola ekskresi cAMP ginjal sebagai respons terhadap pemberian PTH: peningkatan ekskresi cAMP diamati pada tipe 2 dan tidak adanya cAMP pada tipe 1. Diagnosis dipastikan dengan deteksi penurunan kadar nukleotida guanin. pengikatan protein (protein Gs) dalam darah (rata-rata 1,5-2 kali) dibandingkan dengan normalnya. Hipokalsemia biasanya dikombinasikan dengan hiperfosfatemia dan hipofosfaturia. Tingkat PTH meningkat; pada tipe 1C, tingkat PTH normal, sehingga menimbulkan nama “pseudohipoparatiroidisme”. Pemeriksaan sinar-X pada sistem kerangka menunjukkan pemendekan tulang metakarpal dan metatarsal, sering kali demineralisasi umum (osteoporosis), dan penebalan tulang kubah tengkorak. Pola dermatoglif menunjukkan perpindahan aksial palmar triradius.
Kriteria diagnosis:
- bertubuh pendek;
- wajah bulat;
- penundaan gugup perkembangan mental;
- kelainan tulang;
- kalsium serum rendah;
- tingginya kadar hormon paratiroid dalam darah;
- penurunan ekskresi fosfat dan cAMP urin.
PENGOBATAN DAN PENCEGAHAN
Pengobatan hipokalsemia terdiri dari pemberian suplemen kalsium dalam dosis yang cukup untuk mempertahankan konsentrasi kalsium normal dalam darah. Terapi vitamin D sangat penting.Saat ini, metabolit aktif vitamin D digunakan - oksidavit, 1-alpha-D3, kalsitrin, dll dengan dosis 1-2 mcg/hari dengan hasil positif(peningkatan kadar kalsium dalam darah, penurunan gejala sindrom kejang). Tachistin (0,5–1,5 mg/hari) juga efektif. Obat ini meningkatkan penyerapan kalsium di usus dan dengan demikian membantu meningkatkan kadar kalsium dalam darah. Terapi antikonvulsan digunakan sebagai pengobatan tambahan. Pada perkembangan intelektual pengobatan tidak memiliki efek yang nyata, namun seiring dengan penurunan gejala sindrom kejang, regresi diamati manifestasi neurologis(gangguan subkortikal, hiperkinesis koreiform, athetosis, dll). Untuk menghindari overdosis sediaan vitamin D, perlu dilakukan pemantauan konsentrasi kalsium dalam darah setiap 3-7 hari selama 2 minggu pertama pengobatan dan setiap bulan selama 2-3 bulan berikutnya. Setelah konsentrasi kalsium yang stabil dalam darah tercapai, cukup dilakukan pemeriksaan setiap 2-3 bulan sekali. Diet terbatas fosfor membantu menormalkan konsentrasi fosfor dan kalsium dalam darah dan menghilangkan gejala hiperparatiroidisme sekunder. Jika kelenjar endokrin lain tidak mencukupi, terapi penggantian dengan hormon yang sesuai dilakukan.
Pengobatan dengan hormon paratiroid tidak efektif. Untuk meredakan serangan kejang, larutan 10% kalsium klorida atau kalsium glukonat diberikan secara intravena; secara oral – larutan kalsium klorida 5–10%, 1 sendok makan 3–4 kali sehari: kalsium glukonat, kalsium laktat – hingga 10 g per hari.
RAMALAN karena hidup ditentukan oleh tingkat keparahan sindrom kejang.
PENCEGAHAN penyakit ini didasarkan pada data dari konseling genetik medis.
KONSELING MEDIS-GENETIK
Saat melakukan konseling genetik medis, seseorang harus melanjutkan dari jenis pewarisan autosomal dominan dan tingginya (50%) risiko kekambuhan penyakit dalam keluarga dengan bentuk warisan. Untuk mengetahui sifat jenis warisan, perlu dilakukan pemeriksaan menyeluruh terhadap orang tua, karena sindrom ini dapat bermanifestasi secara minimal. gejala klinis. Saat ini, diagnosis genetik molekuler penyakit ini telah dikembangkan dan ditingkatkan dengan mengetikkan mutasi pada gen GNAS1 pada kromosom 20. Metode untuk diagnosis prenatal penyakit secara umum dan tipe individualnya sedang dikembangkan.
OBSERVASI KLINIS Boy G., 14,5 tahun (foto 4), dirawat di Research Clinical Institute of Pediatrics dengan diagnosis: penyakit degeneratif sistem saraf? hidrosefalus eksternal bawaan; epilepsi simtomatik; sindrom keturunan? penyakit penyimpanan? ensefalopati metabolik; hipotiroidisme subklinis; perawakan pendek asal campuran; gangguan kognitif.
Keluhan setelah masuk ke sakit kepala paroksismal intens yang terlokalisasi di daerah frontal dan disertai muntah, yang membawa kelegaan, penurunan daya ingat dan kinerja di sekolah, serangan kejang di mana terjadi kedutan di tangan kanan.
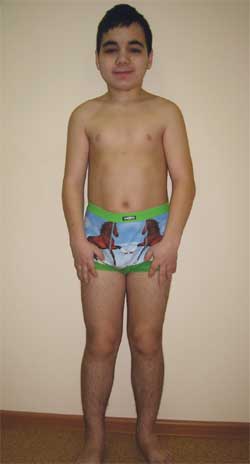
Foto 4.
Anak G., 14,5 tahun, dengan osteodistrofi Albright
(ciri fenotipe, perawakan pendek, anggota tubuh pendek, brachydactyly)
Sejarah keluarga: orang tuanya adalah warga negara Armenia berdasarkan kewarganegaraan, tidak memiliki hubungan darah dan tidak memiliki bahaya pekerjaan. Dalam silsilah kasus penyakit kejiwaan, epilepsi, keterlambatan perkembangan tidak dicatat. Adikku, umur 17 tahun, katanya sehat-sehat saja.
Riwayat hidup dan penyakit: anak laki-laki dari kehamilan ke-2, yang berlangsung tanpa keanehan apa pun, kelahiran kedua, cukup bulan, fisiologis, berat lahir - 3100 g, panjang - 51 cm, langsung menangis, skor Apgar - 7/9 poin. Memburuknya kondisi pada hari ke 3 – kejang neonatal, dihentikan di rumah sakit bersalin. Masa awal pascakelahiran tidak memiliki ciri-ciri. Terjadi sedikit keterlambatan perkembangan motorik pada tahun pertama kehidupan, berjalan mandiri sejak 1 tahun 3 bulan. Sehubungan dengan hal tersebut, beliau diperiksa oleh dokter spesialis saraf dengan diagnosa : lesi organik SSP; hidrosefalus bawaan; kejang neonatal; riwayat kejang demam.
Saya menerima diacarb dan finlepsin. Onset serangan pada umur 1 tahun 11 bulan. – asimetris, tonik dalam bentuk ketegangan tangan kanan dan kaki, dengan mata terbuka, hingga 2 menit, tanpa kehilangan kesadaran, sering hingga 10 episode per hari. Saya menerima Depakine secara tidak teratur. Dengan latar belakang penarikan diri - status tonik tunggal. Pada usia 2 tahun, CT scan otak dilakukan di tempat tinggal, di mana fokus demielinasi tunggal di lobus oksipital diidentifikasi.
Seorang ahli bedah saraf dikonsultasikan dan pengobatan konservatif direkomendasikan. Sejak usia 3 tahun, terjadi keterlambatan perkembangan psiko-bicara, dan dianjurkan observasi oleh psikiater.
Pada usia 4–5 tahun, orang tua mulai memperhatikan adanya deformasi dan pemendekan pada jari tangan dan kaki, terutama jari ke-2–4 yang simetris pada lengan dan tungkai, serta penurunan indikator pertumbuhan. Pada usia 8 tahun, kesimpulan terapis wicara adalah gangguan bicara umum tingkat 2-3; pendidikan di sekolah khusus dianjurkan. Pada usia yang sama, pemeriksaan oleh ahli genetika di tempat tinggal, kesimpulannya: penyakit keturunan menukarkan? Sebuah studi tentang asam amino darah direkomendasikan, tidak ada perubahan yang terdeteksi; kesimpulan akhir: tidak ada bukti penyakit metabolik herediter yang teridentifikasi; hipokondroplasia; Perawatan oleh ahli saraf dan ahli endokrin dianjurkan.
Meja.
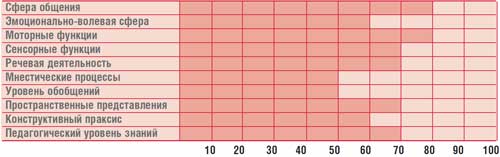
Profil perkembangan mental anak G., 14,5 tahun (IQ = 68)
Pada usia 8 tahun, ia berkonsultasi dengan dokter spesialis endokrinologi mengenai keterlambatan tumbuh kembang. Pemeriksaan rontgen pada tangan menunjukkan ciri-ciri berikut: falang tengah dan utama serta tulang metakarpal memendek dan menebal; Diagnosis ahli radiologi adalah achondroplasia.
Diperiksa berulang kali di tempat tinggal di rumah sakit saraf. Pada usia 12 tahun, serangan kejang muncul tanpa kehilangan kesadaran dengan kedutan pada lengan kanan yang bersifat serial. Terapi antikonvulsan (Depakine) diresepkan, dan frekuensi serangan menurun secara signifikan. Pada usia 13 tahun, MRI otak dengan kontras dilakukan - perubahan simetris pada dasar lobus temporal pada tingkat inti dalam bentuk peningkatan sinyal MR, yang khas untuk racun (mangan) atau ensefalopati metabolik (tembaga, besi).
Diperiksa kembali pada umur 13 tahun 3 bulan. ahli endokrinologi, studi tentang profil tiroid menunjukkan peningkatan hormon perangsang kelenjar gondok(TSH), hipotiroidisme subklinis didiagnosis, L-tiroksin diresepkan.
Saat menganalisis kartu rawat jalan anak dan dokumentasi di tempat tinggal, dilakukan pemeriksaan kalsium dan fosfor sebanyak satu kali, pada usia 1,5 tahun tercatat hipokalsemia, namun tidak dilakukan pemeriksaan lebih lanjut pada kesempatan tersebut. Mengingat ketidakpastian diagnosis di tempat tinggal, ahli genetika mengirim anak tersebut ke Moskow, ke Institut Penelitian Klinis Pediatri Ilmiah, untuk memperjelas diagnosis.
Data penelitian objektif:
Tinggi – 143 cm, berat – 43 kg.
Perkembangan fisik sangat rendah, harmonis, perawakan tidak proporsional akibat pemendekan anggota badan. Pertumbuhan Sds sesuai dengan –2,8 penyimpangan dari norma (norma –2+2).
Ciri-ciri fenotip: wajah bulat, leher pendek, fisura palpebra anti-Mongoloid, batang hidung lebar, dahi tinggi, brachydactyly, pemendekan tulang metakarpal dan metatarsal IV dan V (foto 5). Organ dalam – tanpa kekhasan apa pun. Perkembangan seksual – Tanner tahap III–IV (sesuai dengan usia).
Laboratorium dan studi fungsional:
Analisis klinis darah dan urin normal.
Analisis biokimia darah: kalsium total– 1,39 (normal 2,02–2,6 mmol/l), kalsium terionisasi – 0,61 (normal 1,13–1,32 mmol/l), fosfor anorganik – 3,66 (normal 0,86– 1,56 mmol/l), indikator lainnya dalam batas normal.
Analisis urin biokimia: ekskresi fosfat ginjal berkurang - 11,5 mmol/l (normal 19–32 mmol/l).
Profil tiroid: TSH – 11,75 (kisaran normal 0,4–4,0 µIU/ml), T4 bebas – 0,49 (kisaran normal 1,0–1,8 ng/dL).
Hormon paratiroid – 499 (normal 12–65 pg/ml), STH – 7 ng/ml (normal 7–10 ng/ml), somatomedin-C – 250 ng/ml (normal 88–360 ng/ml).
USG organ dalam- tanpa fitur.
EKG – migrasi alat pacu jantung supraventrikular dengan latar belakang detak jantung reguler 71–80 denyut/menit. Blokade tidak lengkap pada cabang berkas kanan. Pelanggaran proses repolarisasi pada miokardium dinding belakang ventrikel kiri (penurunan z.T III, aVF).
R-grafik tulang belakang – skoliosis sisi kanan dada tulang belakang derajat 1, osteoporosis berat.
R-grafik tangan dengan penangkapan lengan bawah - pemendekan dan pelebaran falang terminal dan tengah. Usia tulang – 13,5–14 tahun.
Tidak ada pola aktivitas epilepsi EEG yang dicatat.
MRI otak - Gambar MR dari beberapa fokus subkortikal dari peningkatan sinyal MR di lobus frontal, hidrosefalus kompensasi eksternal dengan atrofi substansi otak.
MSCT otak menunjukkan area kalsifikasi inti lentiform yang simetris. Area hiperdens difus di thalami, inti kaudat dengan area kalsifikasi di sebelah kanan. Kalsifikasi beberapa titik pada jaringan lunak integumen tengkorak.
Audiogram – tanpa patologi.
Diagnostik DNA pada gen GNAS1 sedang berlangsung.
Konsultasi spesialis:
Ahli endokrinologi – Osteodistrofi herediter Albright tipe 1A (pseudohipoparatiroidisme). Hipotiroidisme primer, kompensasi obat yang tidak lengkap.
Dokter Mata – katarak sekunder lengkap. Direkomendasikan perawatan bedah.
Psikolog – gangguan kognitif (profil psikologis anak disajikan dalam tabel).
Mempertimbangkan fenotipe anak, riwayat kesehatan, hasil penelitian tambahan(hipokalsemia, hiperfosfatemia, hipofosfaturia, peningkatan hormon paratiroid darah), kalsifikasi di otak, adanya katarak, hipotiroidisme), diagnosis ditegakkan: osteodistrofi herediter Albright tipe 1A (pseudohipoparatiroidisme). Disarankan untuk melakukan diagnosa DNA - mencari mutasi pada gen GNAS1.
Perlakuan: Anak dianjurkan mengonsumsi eutirox dengan dosis 100 mcg/hari; metabolit aktif vitamin D - alpha-D3 (Teva) dengan dosis 2 mcg/hari; kalsium (Sandoz) 2000 mg/hari; penggunaan terapi antikonvulsan secara konstan - finlepsin 800 mg/hari di bawah pengawasan ahli saraf-epileptologi; kelas dengan ahli patologi wicara dan psikolog; terapi energi-tropik (Elkar dan koenzim Q10 dalam dosis terkait usia). Memantau indikator metabolisme fosfor-kalsium, kadar hormon paratiroid.
Dengan demikian, Pengamatan klinis yang disajikan menunjukkan kesulitan pencarian diagnostik diferensial, pentingnya studi tepat waktu terhadap parameter biokimia sederhana (dalam kasus epilepsi, skrining berulang indikator metabolisme fosfor-kalsium adalah wajib), hasil dari keterlambatan diagnosis penyakit yang ditentukan secara genetik. , kebutuhan untuk mengintegrasikan tanda-tanda individu ke dalam keseluruhan fenotip penyakit tertentu kondisi patologis untuk diagnosis bentuk individu yang ditargetkan dan tepat waktu penyakit keturunan. Diagnosis tepat waktu dan klarifikasi asal usul setiap sindrom sangatlah penting, karena memungkinkan kita menemukan pendekatan optimal untuk pengobatan kondisi dan pencegahan ini. kemungkinan komplikasi(sampai dengan kecacatan anak); pencegahan terulangnya penyakit keturunan pada keluarga yang terkena dampak (konseling medis dan genetik). Hal ini menentukan perlunya dokter dari berbagai spesialisasi untuk secara jelas menavigasi aliran patologi yang ditentukan secara turun-temurun. Daftar referensi ada di kantor redaksi.